Abrogation of signal-dependent activation of the cdk9/cyclin t2a complex in human rd rhabdomyosarcoma cells
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

_Dear Editor,_ During skeletal myogenesis, the transcription factor MyoD promotes differentiation in myoblasts by its ability to arrest the cell cycle and activate muscle-specific
transcription.1, 2 Recently, we showed that the cdk9/cyclin T2a complex plays an essential role in the MyoD-dependent activation of the myogenic program, describing a distinction with the
cdk1 and cdk2 complexes, which are functionally downregulated during myotube formation and inhibit muscle-specific transcription when overexpressed.3 The role of cdk9 in modulating
promoter-restricted transcription appears to be dependent on the sequence-specific factor recruiting the kinase, and on cdk9's ability to associate with different regulatory subunits,
such as cyclin T1 and cyclin T2.4 _In vitro,_ the cdk9/cyclin T2 complex immunoprecipitated from differentiating myoblasts can efficiently phosphorylate MyoD.3 To further investigate the
relevance of this phosphorylation, we screened a panel of different MyoD point mutants where the serine of putative cdk target sites – proline-directed serine (sp sites) – were replaced by a
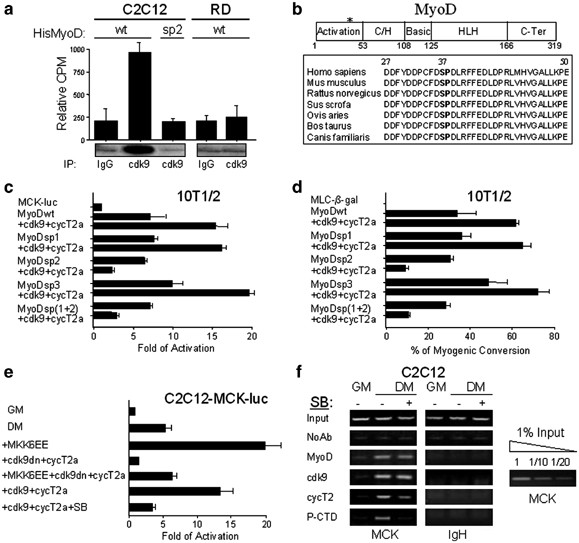
nonphosphorylatable amino acid (alanine). We found that the purified His-MyoD mutant carrying the substitution of serine 37 (His-MyoDsp2) could not be phosphorylated by cdk9 (Figure 1a),
indicating the phosphorylation of this residue as the possible signal enabling cdk9 to transactivate MyoD. To explore this possibility, we decided to compare the transcriptional activity of
MyoDsp2 with two other point mutants carrying a mutation on sp sites targeted by cdk1 and cdk2, serine 5 (MyoDsp1) and serine 200 (MyoDsp3). During the G1 phase of the cell cycle,
cdk2-dependent phosphorylation at serine 200 plays a crucial role in modulating MyoD half-life by triggering the proteosome-dependent degradation pathway.5, 6, 7 In late G2, concomitant
phosphorylation at serine 5 and serine 200 by cdk1 results in the decrease of MyoD activity and stability. In CH310T1/2 fibroblasts induced to differentiate MyoDwt and MyoDsp1 were
efficiently transactivated by cdk9/cyclin T2a, whereas the activity of MyoDsp2 was even impaired by the co-transfection of cdk9/cyclin T2a (Figure 1c). We interpreted this effect as a result
of the formation of a MyoD-cdk9/cyclin T2a complex that cannot be activated by cdk9 phosphorylation, thereby behaving as a dominant-negative toward uncomplexed MyoD. Transactivation of
MyoDsp3 by cdk9/cyclin T2a was similar to that of MyoDwt and MyoDsp1. Finally, we analyzed the function of a MyoD mutant carrying both the mutations in Ser5 and in Ser37 (MyoDsp1+2). This
construct behaved in the same way as MyoDsp2, confirming the possible implication of serine 37 in the transactivation of MyoD by cdk9/cyclin T2a. The same results were obtained by using the
identical MyoD mutants fused to a Gal4 DNA-binding domain (Gal4MyoDwt, sp1, sp2 and sp3) with a Gal4-luciferase construct as a reporter (Supplementary Information, A). As we described
previously, the MyoDNter does not interact with cdk9,3 but includes serine 37. The fact that this mutant is not transactivated by cdk9/cyclin T2a suggests that two regions of MyoD are
required for full activation, the bHLH and serine 37 in the N-terminal transactivation domain. Interestingly, this finding seems to be in accordance with cdk9/cyclin T2's requirement of
two different substrate subdomains, the cdk-phosphorylation site and the recognition/binding site for the cdk/cyclin complex, as for efficiently phosphorylating pRb.8 Moreover, in a
functional test of myogenic conversion, cdk9/cyclin T2a was able to enhance the conversion of fibroblasts into myotubes by ectopic expression of MyoDsp1 and MyoDsp3, like MyoDwt, but failed
to increase the activity of MyoDsp2 and MyoDsp(1+2), which exhibited, again, a reduced myogenic potential when coexpressed with cdk9/cyclin T2a (Figure 1d). These results establish that the
enhancement of MyoD function by cdk9/cyclin T2a requires the presence of serine 37, a well-conserved residue in the MyoD protein of mammals (see Figure 1b). The normal basal activity of the
MyoD serine 37 mutant (MyoDsp2)5 would appear in conflict with the proposed essential function of this serine in receiving an activator signal during muscle differentiation. The complex
regulation of MyoD by a variety of signals and the autoactivatory network of muscle regulatory factors during myogenic differentiation may, at least partially, explain these data. Even when
deficient in receiving one of the positive signals, MyoD can still stimulate a basal level of transcription, which may be enough to activate other myogenic regulatory factors, which can in
turn activate the myogenic program by a parallel cdk9-independent pathway. This explanation is supported by the extremely high redundancy of activation of myogenic transcription in cell
culture and _in vivo_ models.9, 10 Moreover, we cannot rule out that fibroblasts transiently overexpressing MyoD could be deficient for some muscle-specific proteins or signaling pathways,
and ultimately that enforced expression of MyoD could be able to induce trans-differentiation, but not completely recapitulate the myogenic program. The hypothesis that serine 37
phosphorylation could be involved in protein–protein interactions might be suggested by an analogy to the proposed role for MyoD serine 5 in the recruitment of PCAF and HDAC1.11 During the
preparation of this manuscript, Iankova _et al_12 reported that PPAR_γ_ recruits cdk9 complex on specific promoters to activate transcription and stimulate adipogenesis. In accordance with
our model, they showed that cdk9 phosphorylates the adipogenic master regulator at serine 112 to transactivate PPAR_γ_-mediated gene expression. Furthermore, cdk9 can also phosphorylate p53
on serine residues 33, 315 and 392,13 confirming that the kinase complex could recognize signal-dependent transcription factors as additional substrates besides the CTD of RNApolII.4 As the
activity of MyoD is hampered in rhabdomyosarcoma cells, we decided to investigate the significance of cdk9-mediated MyoD phosphorylation in the human cell line RD. For this purpose, we
immunoprecipitated endogenous cdk9 complexes from cells induced to differentiate. Surprisingly, in tumor cells, cdk9 fails to phosphorylate MyoD (Figure 1a), although RD cells showed even
upregulation of both cdk9 and cyclin T2 when compared to C2C12 myoblasts (Supplementary Information, B). Evaluation of immunoprecipitated complexes demonstrated that the antibody could
recognize endogenous cdk9 protein, excluding conformational change of the kinase in cancer cells, and that the amount of precipitated protein was comparable between C2C12 and RD cells
(Supplementary Information, C, upper panel). Further characterization showed that the interaction between cdk9 and cyclin T2 was maintained or even increased, if compared to normal cells
(Supplementary Information, C, lower panel). The paradoxical increase in the protein levels of cdk9/cyclin T2 complex in cancer cells resembled those of some tumor suppressor proteins, such
as p53, whose overexpression often represents a hallmark of gene mutation.14 We explored this possibility by direct sequencing of the DNA coding sequence of cdk9 and cyclin T2 in RD cells,
but we failed to detect any mutation. As endogenous cdk9 kinase activity appeared to be blocked, we verified if enforced expression of cdk9/cyclin T2a could rescue the function of the
complex, but we failed to reach significant values of transcription activation even in the presence of overexpressed MyoD (Supplementary Information, D and E). Our results seem to suggest
that a signaling cascade targeting cdk9 can be inactive in RD cells. Recently, it was reported that RD cells were defective in the MKK6/p38 pathway.15 Moreover, pharmacological blockade of
the p38 pathway reduced the phosphorylation of the CTD of the RNApolII on muscle-specific regulatory regions,16 at residues targeted by cdk9 during transcriptional elongation.17 To test
whether the signal propagated by p38 could affect cdk9 activity in muscle cells, we employed two C2C12 polyclones carrying a chromatin-integrated MyoD-responsive luciferase reporter
controlled, respectively, by the myogenin promoter (myogenin-luc) and the MCK enhancer (MCK-luc). Overexpression of the constitutively active form of the MKK6 kinase (MKK6EE) is able to
strongly coactivate MyoD, inducing even endogenous muscle-specific genes,15, 16, 18 but when co-transfected with the dominant-negative form of cdk9 (cdk9dn), it failed to do so (Figure 1e
and Supplementary Information, F). Significantly, pharmacological blockade of p38 using the specific inhibitor SB203580 abrogated MyoD-dependent transcription, even in the presence of
overexpressed cdk9/cyclin T2a complex (Figure 1e and Supplementary Information, F). These data suggest a functional relationship between cdk9/cyclin T2a and the p38 pathway. We recently
described that p38 signaling influences MyoD-mediated cofactor recruitment on the chromatin of muscle-specific genes,16 and observed that the cdk9/cyclin T2 complex occupies these regions
during the differentiation program.17 To evaluate whether p38 signaling is able to affect cdk9/cyclin T2 localization on the chromatin, we analyzed the presence of cdk9 and cyclin T2 on the
MCK enhancer. We found that even if the binding of cdk9 to the DNA was unaltered, that of cyclin T2 depended, at least to a significant degree, on p38 signaling (Figure 1f). These findings
suggest the hypothesis that a perturbation of the p38 pathway may abrogate the activity of the cdk9/cyclin T2 complex, probably by affecting the stable recruitment of cyclin T2 at the
chromatin level. Upon MKK6-dependent enzymatic activation, p38 localizes on muscle–gene regulatory regions promoting the phosphorylation-dependent recruitment of SWI/SNF, chromatin
remodeling and hyperphosphorylation of the CTD of RNApolII at these elements.16 At least one out of these events could be required for the stabilization of cyclin T2 binding to the DNA.
Moreover, as both cdk9 and cyclin T2 can independently associate with MyoD _in vitro_,3 it is possible that one or more p38-dependent post-translational modifications imparted to the
components of the MyoD transcriptional complex could influence the stable recruitment of the cyclin, even if cdk9 is retained on the chromatin. At present, we cannot exclude that other
mechanisms might be responsible for cdk9/cyclin T2 activation in muscle cells, and we believe it will be important to clarify whether the cdk9/cyclin T2 complex is a direct target of the p38
pathway. Moreover, further studies are awaited to elucidate the significance of cdk9-dependent MyoD phosphorylation in normal and tumor muscle cells _in vivo_. We acknowledge that previous
studies examined the phosphorylation pattern of MyoD without finding a major regulatory function for serine 37.5, 6 However, the phosphotryptic map analyses were performed only in
proliferating myoblasts, and when the transcriptional activity of the MyoDsp2 mutant was tested in differentiating conditions, the authors merely employed luciferase reporter activity assays
in fibroblasts.5 The actual modality of regulation of bHLH-dependent transcription by phosphorylation remains controversial, as numerous studies have reported discrepancies about the role
of different kinases on the regulation of distinct bHLH factors.1 For example, phosphopeptide analysis of MyoD in myoblasts _versus_ myotubes revealed the presence of a
differentiation-induced spot, which corresponds to serine 5 phosphorylation.1 This phosphorylation is abrogated by inactivation of p38 and is ectopically produced in myoblasts by enforced
activation of p38 by MKK6EE.1 Mutation of this residue eliminated the differentiation-induced serine 5 phosphorylation in myotubes, but does not significantly change the activity of MyoD in
fibroblasts.1 Thus, although closely associated with the differentiation process and the activation of p38, the function for this phosphorylation site in myotubes is still unknown. Moreover,
the proposed role for the concomitant phosphorylation at serine 5 and serine 200 by cdk1 in myoblasts, resulting in the repression of MyoD activity and then decrease of its stability,11
seems to be controversial. The presence of a large number of consensus sites for cdks, MAPKs and SAPKs in all the myogenic bHLH proteins would potentially implicate that multiple
phosphorylation signals are required to coordinate muscle-specific transcription. Furthermore, it should be noted that a systematic mutation of all the proline-directed serines in MyoD did
not produce any change of function in fibroblasts, except for serine 200. This scenario shows a complex regulation of MRFs by phosphorylation, and could explain why only co-overexpression of
MyoDsp2 with the cdk9/cyclin T2a complex can demonstrate the role of serine 37 in MyoD transactivation. Similarly, serine 112 of PPAR_γ_ has been reported to be phosphorylated by MAPK, cdk7
and recently by cdk9;12 however, the effects of this covalent modification are so far inconsistent since phosphorylation at the same residue by different kinases results in either
activation or repression. In conclusion, our results establish that cdk9/cyclin T2a-mediated coactivation of MyoD depends on serine 37 phosphorylation. The abrogation of this enzymatic
modification in the human RD cell line suggests the hypothesis that the p38 pathway could represent the upstream cascade signaling to cdk9/cyclin T2 complex in muscle cells. The reported
functional and biochemical links between p53, cdk913 and p38,19 together with the well-known role of these proteins in rhabdomyosarcoma formation,15, 20 corroborate this hypothesis.
REFERENCES * Puri PL, Sartorelli V (2000) _J. Cell. Physiol._ 85: 155–173. * Sartorelli V, Puri PL (2001) _Front. Biosci._ 6: D1024–D1047. * Simone C _et al_. (2002) _Oncogene_ 21:
4137–4148. * Simone C, Giordano A (2001) _Front. Biosci._ 6: D1073–D1082. * Song A _et al_. (1998) _Mol. Cell. Biol._ 18: 4994–4999. * Kitzmann M _et al_. (1999) _Mol. Cell. Biol._ 19:
3167–3176. * Tintignac LA _et al_. (2000) _Exp. Cell Res._ 259: 300–307. * Simone C _et al_. (2002) _Oncogene_ 21: 4158–4165. * Lassar A, Munsterberg A (1994) _Curr. Opin. Cell Biol._ 6:
432–442. * Molkentin JD, Olson EN (1996) _Proc. Natl. Acad. Sci. USA_ 93: 9366–9373. * Tintignac LA _et al_. (2004) _Mol. Cell. Biol._ 24: 1809–1821. * Iankova I _et al_. (2006) _Mol.
Endocrinol_ 20: 1494–1505. * Radhakrishnan SK, Gartel AL (2006) _Cell Cycle_ 5: 519–521. * Chang SC _et al_. (2005) _Int. J. Oncol._ 26: 65–75. * Puri PL _et al_. (2000) _Genes Dev._ 14:
574–584. * Simone C _et al_. (2004) _Nat. Genet._ 36: 738–743. * Giacinti C _et al_. (2006) _J. Cell. Physiol._ 206: 807–813. * Wu Z _et al_. (2000) _Mol. Cell. Biol._ 20: 1809–1821. *
Sanchez-Prieto R _et al_. (2000) _Cancer Res._ 60: 2464–2472. * Merlino G, Helman LJ (1999) _Oncogene_ 18: 5340–5348. Download references ACKNOWLEDGEMENTS We thank Dr. Puri, Dr. Sartorelli
and Dr. Harrington for kindly providing reagents, to Dr. Bagella, Dr. Stiegler and Dr. Bellan for their helpful collaboration, to Dr. Guanti, Dr. Resta and Dr. Stella for critical reading of
the manuscript and to Marie Basso for her editorial assistance. Dr. Simone is supported by ‘Scuola di Specializzazione in Genetica Medica’ from the University of Bari. AUTHOR INFORMATION
AUTHORS AND AFFILIATIONS * Department of Biology, Sbarro Institute for Cancer Research and Molecular Medicine, Center for Biotechnology, College of Science and Technology, Temple University,
1900 N. 12th Street, Bio Life Sciences Building, Room 333, Philadelphia, 19122, PA, USA C Simone & A Giordano * Department of Biomedicine in Childhood, Division of Medical Genetics,
University of Bari, Bari, 70124, Italy C Simone Authors * C Simone View author publications You can also search for this author inPubMed Google Scholar * A Giordano View author publications
You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHORS Correspondence to C Simone or A Giordano. ADDITIONAL INFORMATION Supplementary Information accompanies the
paper on Cell Death and Differentiation website (http://www.nature.com/cdd) SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION A–F (TIF 80 KB) RIGHTS AND PERMISSIONS Reprints and
permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Simone, C., Giordano, A. Abrogation of signal-dependent activation of the cdk9/cyclin T2a complex in human RD rhabdomyosarcoma cells. _Cell
Death Differ_ 14, 192–195 (2007). https://doi.org/10.1038/sj.cdd.4402008 Download citation * Published: 14 July 2006 * Issue Date: 01 January 2007 * DOI:
https://doi.org/10.1038/sj.cdd.4402008 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative