Brostallicin (pnu-166196) – a new dna minor groove binder that retains sensitivity in dna mismatch repair-deficient tumour cells
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Defects in DNA mismatch repair (MMR) are associated with a predisposition to tumorigenesis and with drug resistance owing to high mutation rates and failure to engage
DNA-damage-induced apoptosis. DNA minor groove binders (MGBs) are a class of anticancer agents highly effective in a variety of human cancers. Owing to their mode of action, DNA MGB-induced
DNA damage may be a substrate for DNA MMR. This study was aimed at investigating the effect of loss of MMR on the sensitivity to brostallicin (PNU-166196), a novel synthetic
_α_-bromoacrylic, second-generation DNA MGB currently in Phase II clinical trials and structurally related to distamycin A. Brostallicin activity was compared to a benzoyl mustard derivative
of distamycin A (tallimustine). We report that the sensitivities of MLH1-deficient and -proficient HCT116 human colon carcinoma cells were comparable after treatment with brostallicin,
while tallimustine resulted in a three times lower cytotoxicity in MLH1-deficient than in -proficient cells. MSH2-deficient HEC59 parental endometrial adenocarcinoma cells were as sensitive
as the proficient HEC59+ch2 cells after brostallicin treatment, but were 1.8-fold resistant after tallimustine treatment as compared to the MSH2-proficient HEC59+ch2 counterpart. In
addition, p53-deficient mouse fibroblasts lacking PMS2 were as sensitive to brostallicin as PMS2-proficient cells, but were 1.6-fold resistant to tallimustine. Loss of neither ATM nor DNA-PK
affected sensitivity to brostallicin in p53-deficient mouse embryonic fibroblasts, indicating that brostallicin-induced cytotoxicity in a p53-deficient genetic background does not seem to
require these kinases. These data show that, unlike other DNA MGBs, MMR-deficient cells retain their sensitivity to this new _α_-bromoacrylic derivative, indicating that brostallicin-induced
cytotoxicity does not depend on functional DNA MMR. Since DNA MMR deficiency is common in numerous types of tumours, brostallicin potentially offers the advantage of being effective against
MMR-defective tumours that are refractory to several anticancer agents. SIMILAR CONTENT BEING VIEWED BY OTHERS MOLECULAR DISRUPTION OF DNA POLYMERASE Β FOR PLATINUM SENSITISATION AND
SYNTHETIC LETHALITY IN EPITHELIAL OVARIAN CANCERS Article Open access 05 March 2021 ALTERNATE THERAPEUTIC PATHWAYS FOR PARP INHIBITORS AND POTENTIAL MECHANISMS OF RESISTANCE Article Open
access 25 January 2021 IN SILICO SCREENING IDENTIFIES A NOVEL SMALL MOLECULE INHIBITOR THAT COUNTERACTS PARP INHIBITOR RESISTANCE IN OVARIAN CANCER Article Open access 13 April 2021 MAIN
Minor groove binders (MGBs) represent an interesting class of anticancer agents, which have been shown to be highly effective in _in vitro_ and _in vivo_ preclinical tumour models
unresponsive to other antineoplastic agents (Martin et al, 1981; Li et al, 1982, 1992; Hartley et al, 1988; D'Alessio et al, 1994; D'Incalci, 1994; Colella et al, 1999; Marchini et
al, 1999; Geroni et al, 2002). The main representatives of this class, which reached the clinic, are the antitumour agents derived from CC-1065, that is, adozelesin, carzelesin, and
bizelesin, and the distamycin A derivative tallimustine. These ‘classical’ MGBs have been shown to be highly DNA sequence-specific (Lee et al, 1993; D'Incalci, 1994) and to exert their
cytotoxic effect through the ability to _per se_ directly alkylate DNA mainly at the N3 position of adenines exposed in (TA)-rich sequences in the DNA minor groove (Hurley et al, 1984;
Reynolds et al, 1985; Broggini et al, 1995, 1991; Sun and Hurley, 1992; D'Incalci, 1994; Marchini et al, 1998), without the requirement to be activated by other pathways (e.g.,
enzymatic activation of the drug). The absence of significant antitumour activity for nonalkylating MGBs (Marchini et al, 1998) indicates that the N3 alkylation activity of these compounds
is a prerequisite for their cytotoxicity. MGBs activity, however, has previously been reported (Colella et al, 1999) to be associated with reduced susceptibility to the cytotoxic effect in
tumour cells with defects in DNA mismatch repair (MMR), similar to certain chemicals, including MNNG, which alkylates O6 of guanines, and anticancer agents such as doxorubicin and cisplatin
(Branch et al, 1995; Drummond et al, 1996). MMR proteins recognise mismatched base pairs in the DNA, arising either spontaneously during DNA metabolism or from modified nucleotides provoked
by physical and chemical agents, and are thought to link DNA damage recognition to an apoptotic pathway, thereby preventing mutagenesis, tumorigenesis, and tumour progression (Modrich, 1991;
Fink et al, 1998). Tumours resulting from MMR-deficiency include the hereditary nonpolyposis colon cancer (HNPCC) and some sporadic carcinomas such as mammary, ovarian, or endometrial
cancers (Peltomaki, 2001). The development of novel MGBs able to overcome the involvement of MMR assumes great clinical importance with respect to the treatment of tumours deficient in MMR.
A novel _α_-bromoacryloyl derivative of distamycin A, PNU-151807, which exhibits no alkylating activity _per se_, has been identified (Marchini et al, 1999). The cytotoxic effect has been
shown to not depend on MLH1 in some tumour cells (Colella et al, 1999) and has been attributed to the _α_-bromoacrylic moiety of the compound, which seems to interfere with cell cycle
progression via yet unknown pathways (Cozzi, 2000; Geroni et al, 2002). Recently, brostallicin (PNU-166196), a synthetic _α_-bromoacrylic MGB structurally related to PNU-151807, has been
selected for clinical development. Brostallicin has shown very promising activity in experimental tumour models; its _in vitro_ and _in vivo_ activity is increased in tumour cells with
higher glutathione (GSH) and/or glutathione-_S_-transferase (GST) levels (Geroni et al, 2002). The _α_-bromoacrylic moiety of brostallicin was found to react with GSH, in a reaction
catalysed by GST, with the possible formation of a highly reactive GSH-complex able to bind covalently to DNA (Geroni et al, 2002; Cozzi, 2003). The present study was aimed at investigating
the effect of loss of MMR on the sensitivity to brostallicin compared to the structurally related tallimustine, using cell lines deficient or proficient in MLH1, MSH2, or PMS2, respectively.
A putative involvement of two members of the PI3-like kinase family, ATM and DNA-PK, which link DNA damage and cell cycle response in drug-induced cytotoxicity, was also investigated. We
report that MMR-deficient cells retain sensitivity to brostallicin, thereby extending the list of potential anticancer agents for use in the treatment of MMR-deficient tumours, and that
brostallicin-induced cytotoxicity may not require ATM and DNA-PK. MATERIALS AND METHODS CELL LINES The MLH1-deficient human colorectal adenocarcinoma cell line HCT116 was obtained from the
American Type Culture Collection (ATCC CCL 247), and a subline complemented with chromosome 3 carrying the wild-type gene for _hMLH1_ (clone HCT116/3–6, identified here as HCT116+ch3) was
obtained from Dr M Koi (Koi et al, 1994) as were the MSH2-deficient human endometrial adenocarcinoma cell line HEC59 (Umar et al, 1997) and a subline complemented with chromosome 2 carrying
the wild-type gene for _hMSH2_ (clone HEC59/2–4, identified here as HEC59+ch2). HCT116 cells contain a hemizygous mutation in _MLH1_ resulting in a truncated, nonfunctional protein (Boyer et
al, 1995). Similarly, the HEC59 cells are mutated at different loci on both alleles of _MSH2_ and are deficient in repair activity (Umar et al, 1997). The chromosome-complemented sublines
HCT116+ch3 and HEC59+ch2 are competent in DNA MMR. HCT116 and HEC59 cell lines were maintained in Iscove's modified Dulbecco's medium (Life Technologies, Basel, Switzerland)
supplemented with 2 mM L-glutamine and 10% heat-inactivated foetal bovine serum (Oxoid, Basel, Switzerland). The chromosome-complemented lines were maintained in a medium supplemented with
geneticin (400 _μ_g ml−1 for HCT116+ch3, and 600 _μ_g ml−1 for HEC59+ch2) (Life Technologies). Although the extent of possible effects of the introduction of an extra chromosome are not
fully clear, it is generally acknowledged that it does not spoil the effects of loss of MMR on drug sensitivity. PMS2−/−/p53−/− and PMS2+/+/p53−/− cell lines, established from
E1A/Ha-Ras-transformed knockout mice primary fibroblasts, were generously provided by Dr P Glazer (Zeng et al, 2000). Cells are maintained in culture for a limited number of passages and are
routinely tested for the expression of MMR proteins. The ATM+/+/p53−/− and ATM−/−/p53−/−, and the DNA-PK+/+/p53−/− and DNA-PK−/−/p53−/− mouse embryonic fibroblasts, were generously provided
by Dr P Leder (Westphal et al, 1997) and Dr EH Goodwin (Bailey et al, 1999), respectively. The cells were maintained in DMEM medium supplemented with 2 mM L-glutamine (Life Technologies),
10% heat-inactivated foetal calf serum (Oxoid) and penicillin/streptomycin (100 U ml−1/100 _μ_g ml−1, Life Technologies). All cell lines were tested negative for contamination with
_Mycoplasma_ spp. and maintained in a controlled environment of 5% CO2 and 95% relative humidity at 37°C. Except for the ATM+/+/p53−/− and ATM−/−/p53−/− mouse cells, which grow as a
monolayer and do not form colonies, all other cell lines used in these experiments form well-defined individual colonies when seeded sparsely on standard tissue culture plates. REAGENTS
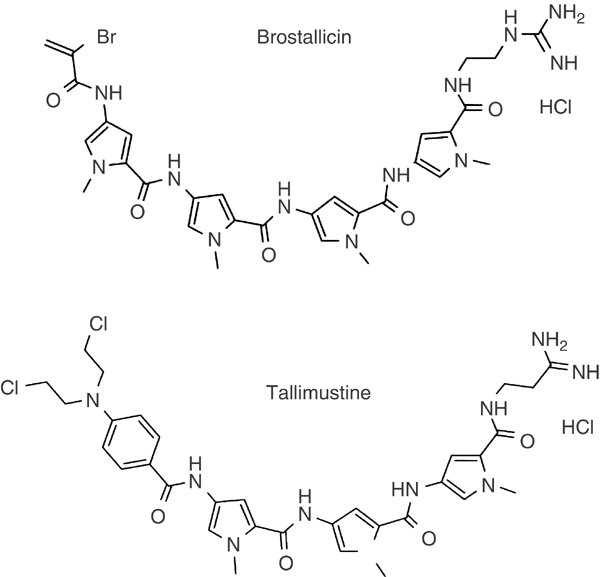
Distamycin A and its derivatives brostallicin (PNU-166196) and tallimustine (PNU-152241) were synthesised by Pharmacia Italy (Nerviano, Italy). The chemical structures of the derivatives are
presented in Figure 1. Brostallicin was dissolved in methanol, tallimustine in DMSO, and distamycin A in water. Stock solutions were stored at −20°C. The final concentration of DMSO or
methanol in the cultures was <0.1% at all drug concentrations and in controls. Previous experiments (data not shown) have shown that neither 0.1% DMSO nor 0.1% methanol affects the
viability or growth of these cell lines. MPE FOOTPRINTING ANALYSIS The MPE footprinting method has been previously described in detail (Hertzberg and Dervan, 1984). The 4492- and 751-bp
fragments of SV40-labelled plasmid previously described (Marchini et al, 1999) were incubated with distamycin A, tallimustine, and brostalicin (50 _μ_ M) for 1 h at room temperature and
treated for 30 min at room temperature with a solution of MPE-(NH4)2-Fe(SO4)2 (synthesised by Pharmacia, Italy, according to the published method; Hertzberg and Dervan, 1984). After
precipitation, DNA was resuspended in loading buffer and electrophoresed on an 8% polyacrylamide 7 M urea gel and autoradiographed. TAQ POLYMERASE STOP ASSAY Studies with the Taq stop assay
were based on a previously reported method (Ponti et al, 1991). Prior to drug-DNA incubation, plasmid pBSSK-TOPO II was linearised with a _Pst_I restriction enzyme (NEB) to provide a stop
for the Taq polymerase, downstream from the primer. After drug treatment, the DNA was precipitated and washed as described (Ponti et al, 1991). The primer was 5′-end labelled with T4
polynucleotide kinase (NEB) and [_γ_-32P] ATP (5000 Ci mmol−1, Amersham). The synthetic primer sequence and the linear PCR amplification conditions were performed as described (Marchini et
al, 1998). Samples were then purified by extraction with an equal volume of chloroform–isoamyl alcohol (24 : 1), and then precipitated and washed following the standard protocol. Dried
samples were resuspended in loading buffer and denatured at 90°C for 2 min before loading onto an 8% polyacrylamide denatured gel. After the run, the gel was dried and autoradiographed.
CLONOGENIC SURVIVAL AND MTT PROLIFERATION ASSAYS Clonogenic survival in response to drug treatment was performed by plating 250 cells in 60 mm cell culture dishes. After 24 h, the drug was
added, followed by incubation in a drug-containing medium for 2 h or 24 h and then in a drug-free medium for another 6–8 days at 37°C in a humidified atmosphere containing 5% carbon dioxide.
Cells were then fixed with 25% acetic acid in ethanol and stained with Giemsa. Colonies of at least 50 cells were scored visually. Each experiment was performed a minimum of three times
using triplicate cultures for each drug concentration. The logarithm of relative colony formation was plotted against the concentration of the drug. The IC50 was estimated by linear
interpolation of the logarithmic transformed relative plating efficiencies. For ATM+/+/p53−/− and ATM−/−/p53−/− mouse cells that do not form distinct colonies, the drug sensitivity was
determined by the MTT assay (Mosmann, 1983). MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazoliumbromide) measures the mitochondrial dehydrogenase of surviving cells. Cells growing
in the log phase were harvested by brief trypsinisation. A total of 1000 cells were plated (96 well plates) 24 h prior to 2 h drug treatment. Cells were then grown in a drug-free medium for
another 4 days at 37°C in a humidified atmosphere containing 5% carbon dioxide. A volume of 20 _μ_l MTT in PBS to a final concentration of 0.5 mg ml−1 was added, followed by incubation at
37°C for 4 h, aspiration of the medium, and addition of 200 _μ_l DMSO. The optical density was measured by the _E_max microplate reader E9336 (Molecular Devices, Clearwater, MN, USA) at 540
nm, setting the value of the cell lines in the medium to 1.0 (control) and the value of the no cells blank to zero. Differences in drug sensitivity of the respective cell lines were
determined from at least four independent experiments and are reported as the concentration required to suppress proliferation by 50% (IC50). STATISTICAL ANALYSIS The mean±s.d. values were
calculated for all data sets. The two-sided paired _t_-test was used to compare the effects on drug sensitivity. _P_<0.05 was considered to be statistically significant. RESULTS
BROSTALLICIN DOES NOT ALKYLATE DNA _PER SE_ BUT THROUGH THE INTERACTION WITH GSH/GST Noncovalent interactions of brostallicin and tallimustine (TAM) with DNA were compared to those of
distamycin A (DISTA). The data reported in Figure 2 show an autoradiograph of a classical ladder of an MPE-footprinting experiment tested on the 751-bp (panel A) and 4492-bp fragments (panel
B) of the SV40 DNA plasmid. Each band corresponds to a DNA fragment differing in size by a single nucleotide. When a DNA region is protected by the presence of a molecule, chemical
digestion is blocked and a ‘gap’ is present on the autoradiograph. In the control (CTR, Figure 2), all fragments are present with broadly the same signal intensity on the gel, while in all
the treated sample lanes a typical ‘gap’ is common in AT-rich regions. The brackets highlight these regions. The distamycin A backbone present in the brostallicin chemical structure drives
the DNA interaction towards AT-rich regions in the same way as previously shown for tallimustine. In fact, brostallicin shows a noncovalent DNA interaction effect superimposable to that of
tallimustine and distamycin A (internal positive control). These regions are highlighted by brackets. The differences in band intensities were due to differences in gel loading. On the basis
of the previously reported data showing that brostallicin is able to covalently interact with DNA upon _in vitro_ reduction by the GSH/GST system (Geroni et al, 2002), we further tested
this hypothesis by incubating the drug-DNA solution with and without GSH and GST in an _in vitro_ system. The sequence-specific, covalent DNA interaction of brostallicin in comparison with
tallimustine was analysed by the Taq polymerase stop assay. This assay is a linear amplification method employing the properties of DNA polymerase to investigate the sequence selectivity of
the interaction between DNA-damaging agents and the DNA. As expected, tallimustine retained its high sequence specificity in alkylating DNA at the target hexamer (5′-TTTTGA), while
brostallicin _per se_ was completely unable to produce any alkylation in the selected DNA region (Figure 3). Brostallicin alone did not alkylate DNA, while a band was observed when GST/GSH
was added to the reaction mixture. In the absence of brostallicin, GSH/GST did not induce any alteration able to block Taq polymerase. It is important to underline the fact that, although
the interaction of brostallicin with DNA involves AT-rich regions, the compound binds to a sequence (AAAG) different from those previously reported for tallimustine. Studies are still in
progress to better define the sequence of the alkylated regions. LOSS OF MLH1 OR MSH2 DOES NOT ALTER SENSITIVITY TO BROSTALLICIN The question was addressed as to whether loss of either MLH1
or MSH2 affects the sensitivity to brostallicin using the clonogenic assay. The data presented in Table 1 show that MLH1-deficient HCT116 cells are nearly as sensitive as MLH1-proficient
HCT116+ch3 cells to this drug (_P_=0.36). This indicates that MLH1 is not involved in brostallicin-mediated cytotoxicity. Furthermore, MSH2-deficient HEC59 cells are as sensitive to
brostallicin as MSH2-proficient HEC59+ch2 cells (_P_=0.41), indicating that brostallicin-mediated cytotoxicity does not require functional MSH2. Brostallicin cytotoxiciy has been compared to
tallimustine. The results show that MLH1-deficient and MSH2-deficient cells are three-fold (_P_<0.01) and 1.8-fold (_P_=0.03), respectively, less sensitive to tallimustine than their
respective proficient counterparts. SENSITIVITY TO BROSTALLICIN, BUT NOT TO TALLIMUSTINE, IS RETAINED AFTER LOSS OF PMS2 Although less frequently mutated than MLH1 or MSH2 in human cancers,
PMS2 may nevertheless be relevant in this respect since it forms a heterodimer with MLH1 and the lack of one or the other partner affects MMR activity. Based on the model that cytotoxicity
of tallimustine, but not the _α_-bromoacrylic derivatives, is dependent on functional MMR, it is anticipated that loss of PMS2 negatively affects sensitivity to tallimustine, but not to
brostallicin. The effect of loss of PMS2 on drug sensitivity was investigated in p53-deficient cells derived from knockout mice. Table 2 shows that the clonogenic survival after treatment
with brostallicin in PMS2-deficient cells was not different from that in PMS2-proficient cells (_P_=0.79). In contrast, PMS2-deficient cells were 1.6-fold less sensitive to tallimustine than
PMS2-proficient cells (_P_=0.02). Thus, PMS2-deficient p53-null mouse fibroblasts retain sensitivity to brostallicin. The 1.6-fold resistance to tallimustine in PMS2-deficient cells
indicates a role for PMS2 in sensitivity to this compound. LOSS OF ATM OR DNA-PK DOES NOT AFFECT SENSITIVITY TO BROSTALLICIN It has previously been proposed that the cytotoxic effect of the
_α_-bromoacrylic derivative PNU-151807 interferes with the cell cycle checkpoint control (Marchini et al, 1999). Although yet unknown, a possible pathway may include ATM or DNA-PK, members
of the PI3-like kinase family, which are important kinases for connecting DNA damage monitoring and cellular responses such as cell cycle checkpoint activation and apoptosis. The question
was addressed as to whether the sensitivity to brostallicin is affected by loss of ATM or DNA-PK in a p53-deficient genetic background. We used embryonic fibroblasts from knockout mice. The
data presented in Figure 4 show that ATM-deficient cells (0.8±0.3 _μ_ M) were as sensitive to brostallicin as ATM-proficient cells (0.9±0.2 _μ_ M) in a p53-deficient genetic setting
(_P_=0.60). Likewise, DNA-PK-deficient cells (17.5±0.7 nM) were as sensitive to brostallicin as DNA-PK-proficient cells (21.0±1.4 nM) in a p53-null background (_P_=0.13). Thus, neither ATM
nor DNA-PK seems to be involved in the sensitivity to brostallicin in p53-deficient mouse cells. DISCUSSION The present study demonstrates that brostallicin, a novel _α_-bromoacrylic,
second-generation DNA MGB structurally related to distamycin A, maintains its cytotoxic effect in cells deficient for the MMR proteins MLH1, MSH2, or PMS2. The data permit drawing several
conclusions. First, brostallicin, the lead compound of a novel class of MGBs in clinical trials, exerts its cytotoxic effect regardless of the MMR status, suggesting that further clinical
testing of brostallicin in tumours deficient in MMR is to be recommended. Second, brostallicin-induced cytotoxicity can occur in the absence of functional ATM or DNA-PK in p53-deficient
cells, indicating that brostallicin-induced cytotoxicity in this setting is independent of PI3-like kinase family status. Third, brostallicin is the first MGB unable to _per se_ covalently
interact with DNA. It requires the GSH/GST system to alkylate DNA with a sequence specificity different from that reported for previously tested alkylating molecules. MMR plays an important
role in the correction of spontaneously occurring errors during DNA processing that have escaped the DNA polymerase proof-reading activity, thereby preserving the integrity of the genome by
preventing the occurrence of gene mutations and tumorigenesis (Modrich, 1991). Spontaneous tumours arising from MMR deficiency include the hereditary nonpolyposis colon cancer as well as
some sporadic carcinomas such as mammary, ovarian, or endometrial cancers (Peltomaki, 2001). MMR monitors specific types of DNA damage introduced by DNA-damaging agents, and subsequently
triggers an apoptotic response (Fink et al, 1998). Loss of MMR hence results in resistance to a variety of widely used anticancer drugs, including the topoisomerase I poisons camptothecin
and topotecan, the topoisomerase II poisons doxorubicin, epirubicin, mitoxantrone and etoposide, and some platinum compounds such as cisplatin and carboplatin, as well as some alkylating
agents including MNNG and busulphan (Branch et al, 1995; Drummond et al, 1996; Fink et al, 1998, 1996; Fedier et al, 2001). Interestingly, the MMR status also affects the activity of several
MGBs such as CC-1065 analogues and the distamycin-derivative tallimustine, but not that of the _α_-bromoacryoyl derivative of distamycin A (PNU-151807) (Colella et al, 1999). The present
study expands on this previous finding by demonstrating that brostallicin, a novel second-generation DNA MGB structurally related to PNU-151807, exerts its cytotoxic effect regardless of the
MMR status. Brostallicin as well as the class of the taxanes (Fedier et al, 2001) and photodynamic therapy (Schwarz et al, 2002) may thus represent valuable options for the treatment of
tumours disabled in MMR. The _α_-bromoacryoyl moiety has been proposed to be important since it reacts with GSH, and reactive drug-GSH intermediates may then modify the DNA by mechanisms not
yet fully understood (Geroni et al, 2002; Cozzi, 2003). DNA interaction data reported in the present study suggest that the distamycin A backbone drives the brostallicin molecule towards
the AT regions present in the minor groove of the DNA. In addition, brostallicin binds covalently to DNA through interaction with the GSH/GST system. Brostallicin covalently binds to DNA
with a completely different sequence specificity than tallimustine. One hypothesis for the different behaviour of brostallicin against MMR status is that this covalent interaction is not
substrate for MMR, whereas the alkylated DNA by tallimustine is recognised by MMR. It should be noted that no direct interaction between MMR and the GSH/GST system is known, and that the
GSH/GST status of the cell lines under study does not matter for the experiments because the cell lines are quasi-isogenic, that is, they differ only in their MMR status and the extra
chromosomes. Moreover, as reported for PNU-151807, the bromoacryloyl moiety seems to be relevant for cell cycle checkpoint control (Marchini et al, 1999). The identity of mediators for
signalling between DNA damage and downstream effectors is not clear. One possibility is that the DNA damage is recognised by one or several members of BASC (BRCA1-associated genome
surveillance complex), a multiprotein complex including BRCA1, ATM, MMR proteins, and other proteins implicated in DNA repair (Wang et al, 2000). Our data, however, show that deficiency in
ATM or DNA-PK did not affect brostallicin sensitivity in p53-deficient cells, arguing against a role of these kinases in these cells. Since these kinases are activated upon DNA double-strand
breaks introduced by radiation or radiomimetic drugs (Jackson, 1997; Smith et al, 1999), _α_-bromoacryoyl derivatives seem unlikely to produce this type of lesion. Although the cytotoxic
effect of tallimustine and PNU-151807 has been shown not to be dependent on the p53 status (Marchini et al, 1999), the data for these kinases obtained in p53-deficient cells may not be
conclusive for p53-proficient cells. There is an apparent higher sensitivity to brostallicin of the DNA-PK data set compared to the ATM data set, but this is likely due to the use of two
assays that differ in their sensitivities. Mutations in the p53 tumour suppressor gene are found in a large fraction of human cancers (Hollstein et al, 1991) and this may be the genetic
basis underlying failure to respond to chemotherapy (Ferreira et al, 1999). PNU-151807 has recently been reported to retain sensitivity against cells disabled in p53 function (Marchini et
al, 1999), indicating that PNU-151807-mediated cytotoxicity does not require functional p53. We have recently shown that additional loss of PMS2 in p53-deficient cells increases cytotoxicity
to a variety of anticancer agents (Fedier et al, 2002). This hypersensitising effect, however, was not observed in response to treatment with brostallicin. For tallimustine, even an
opposite effect was observed in PMS2-deficient cells, suggesting that tallimustine-induced DNA damage is a substrate for MMR in p53-deficient cells. Consistent with this,
tallimustine-induced DNA damage has already been shown to be a substrate for MMR in p53-proficient cells (Colella et al, 1999). We also observed that tallimustine is less toxic than
brostallicin in p53-deficient cells and that this effect is much greater than the difference in sensitivity to tallimustine between MMR-deficient and -proficient cells. This marked effect
was not observed in p53-proficient cells. As the status of p53 has been reported not to markedly affect the sensitivity of human tumour cells to either tallimustine or PNU-151807 (Marchini
et al, 1998), this effect in p53-deficient cells may be ascribed to the mouse origin and/or to the fibroblast cell type. In summary, the present study demonstrates that brostallicin-mediated
cytotoxicity does not depend on the MMR status of tumour cells, and that, at least in p53-deficient mouse cells, functional ATM or DNA-PK is not required. Brostallicin potentially offers
the advantage of having efficacy on MMR-defective tumours that are refractory to several anticancer agents. Since the responsiveness to cisplatin treatment is affected by both MMR status and
GSH/GST level/expression, brostallicin is a good candidate for clinical protocols. CHANGE HISTORY * _ 16 NOVEMBER 2011 This paper was modified 12 months after initial publication to switch
to Creative Commons licence terms, as noted at publication _ REFERENCES * Bailey SM, Meyne J, Chen DJ, Kurimasa A, Li GC, Lehnert BE, Goodwin EH (1999) DNA double-strand break repair
proteins are required to cap the ends of mammalian chromosomes. _Proc Natl Acad Sci USA_ 96: 14899–14904 Article CAS PubMed PubMed Central Google Scholar * Boyer JC, Umar A, Risinger
JI, Lipford JR, Kane M, Yin S, Barrett JC, Kolodner RD, Kunkel TA (1995) Microsatellite instability, mismatch repair deficiency, and genetic defects in human cancer cell lines. _Cancer Res_
55: 6063–6070 CAS PubMed Google Scholar * Branch P, Hampson R, Karran P (1995) DNA mismatch binding defects, DNA damage tolerance, and mutator phenotypes in human colorectal carcinoma
cell lines. _Cancer Res_ 55: 2304–2309 CAS PubMed Google Scholar * Broggini M, Coley HM, Mongelli N, Pesenti E, Wyatt MD, Hartley JA, D'Incalci M (1995) DNA sequence-specific adenine
alkylation by the novel antitumor drug tallimustine (FCE 24517), a benzoyl nitrogen mustard derivative of distamycin. _Nucleic Acids Res_ 23: 81–87 Article CAS PubMed PubMed Central
Google Scholar * Broggini M, Erba E, Ponti M, Ballinari D, Geroni C, Spreafico F, D'Incalci M (1991) Selective DNA interaction of the novel distamycin derivative FCE 24517. _Cancer
Res_ 51: 199–204 CAS PubMed Google Scholar * Colella G, Marchini S, D'Incalci M, Brown R, Broggini M (1999) Mismatch repair deficiency is associated with resistance to DNA minor
groove alkylating agents. _Br J Cancer_ 80: 338–343 Article CAS PubMed PubMed Central Google Scholar * Cozzi P (2000) Recent outcome in the field of distamycin-derived minor groove
binders. _Farmaco_ 55: 168–173 Article CAS PubMed Google Scholar * Cozzi P (2003) The discovery of a new potential anticancer drug: a case history. _Farmaco_ 58: 213–220 Article CAS
PubMed Google Scholar * D'Alessio R, Geroni C, Biasoli G, Pesenti E, Grandi M, Mongelli N (1994) Structure–activity relationship of novel distamycin A derivatives: synthesis and
antitumor activity. _Bioorganic Med Chem Lett_ 4: 1467–1472 Article CAS Google Scholar * D'Incalci M (1994) DNA-minor groove alkylators, a new class of anticancer agents. _Ann Oncol_
5: 877–878 Article CAS PubMed Google Scholar * Drummond JT, Anthoney DA, Brown R, Modrich P (1996) Cisplatin and adriamycin resistance are associated with MutL_α_ and mismatch repair
deficiency in an ovarian tumor cell line. _J Biol Chem_ 271: 19645–19648 Article CAS PubMed Google Scholar * Fedier A, Ruefenacht UB, Schwarz VA, Haller U, Fink D (2002) Increased
sensitivity of p53-deficient cells to anticancer agents due to loss of Pms2. _Br J Cancer_ 87: 1027–1033 Article CAS PubMed PubMed Central Google Scholar * Fedier A, Schwarz VA, Walt H,
Delli Carpini R, Haller U, Fink D (2001) Resistance to topoisomerase poisons due to loss of DNA mismatch repair. _Int J Cancer_ 93: 571–576 Article PubMed Google Scholar * Ferreira CG,
Tolis C, Giaccone G (1999) p53 and chemosensitivity. _Ann Oncol_ 10: 1011–1021 Article CAS PubMed Google Scholar * Fink D, Aebi S, Howell SB (1998) The role of DNA mismatch repair in
drug resistance. _Clin Cancer Res_ 4: 1–6 CAS PubMed Google Scholar * Fink D, Nebel S, Aebi S, Zheng H, Cenni B, Nehme A, Christen RD, Howell SB (1996) The role of DNA mismatch repair in
platinum drug resistance. _Cancer Res_ 56: 4881–4886 CAS PubMed Google Scholar * Geroni C, Marchini S, Cozzi P, Galliera E, Ragg E, Colombo T, Battaglia R, Howard M, D'Incalci M,
Broggini M (2002) Brostallicin, a novel anticancer agent whose activity is enhanced upon binding to gluthathione. _Cancer Res_ 62: 2332–2336 CAS PubMed Google Scholar * Hartley JA, Lown
JW, Mattes WB, Kohn KW (1988) DNA sequence specificity of antitumor agents. Oncogenes as possible targets for cancer therapy. _Acta Oncol_ 27: 503–510 Article CAS PubMed Google Scholar *
Hertzberg RP, Dervan PB (1984) Cleavage of DNA with methidiumpropyl-EDTA-iron(II): reaction conditions and product analyses. _Biochemistry_ 14: 3934–3945 Article Google Scholar *
Hollstein M, Sidransky B, Vogelstein B, Harris CC (1991) p53 mutations in human cancers. _Science_ 253: 49–53 Article CAS PubMed Google Scholar * Hurley LH, Reynolds VL, Swenson DH,
Petzold GL, Scahill TA (1984) Reaction of the antitumor antibiotic CC-1065 with DNA: structure of a DNA adduct with sequence specificity. _Science_ 226: 843–844 Article CAS PubMed Google
Scholar * Jackson SP (1997) DNA-dependent protein kinase. _Int J Biochem Cell Biol_ 29: 935–938 Article CAS PubMed Google Scholar * Koi M, Umar A, Chauhan DP, Cherian SP, Carethers JM,
Kunkel TA, Boland CR (1994) Human chromosome 3 corrects mismatch repair deficiency and microsatellite instability and reduces _N_-methyl-_N_′-nitro-_N_-nitrosoguanidine tolerance in colon
tumor cells with homozygous hMLH1 mutation. _Cancer Res_ 54: 4308–4312 CAS PubMed Google Scholar * Lee M, Rhodens VL, Wyatt MD, Forrow S, Hartley JA (1993) Design, synthesis, and
biological evaluation of DNA sequence and minor groove selective alkylating agents. _Anticancer Drug Des_ 8: 173–193 CAS PubMed Google Scholar * Li LH, Dekonong TF, Kelly RC, Krueger WC,
McGovren JP, Padbury GE, Petzold GL, Wallace TL, Ouding RJ, Prairie MD, Gebhard I (1992) Cytotoxicity and antitumor activity of carzelesin, a prodrug cyclopropylpyrroloindole analogue.
_Cancer Res_ 52: 4904–4913 CAS PubMed Google Scholar * Li LH, Swenson D, Schpock S, Kuentzel S, Dayton B, Kreiger W (1982) CC-1065 (NSC-298223) a novel antitumor agent that interacts
strongly with double-strand DNA. _Cancer Res_ 42: 999–1104 CAS PubMed Google Scholar * Marchini S, Ciro M, Gallinari F, Cozzi P, D'Incalci M, Broggini M (1999) Bromoacryloyl
derivative of distamycin A (PNU 151807): a new non-covalent minor groove DNA binder with antineoplastic activity. _Br J Cancer_ 80: 991–997 Article CAS PubMed PubMed Central Google
Scholar * Marchini S, Cozzi P, Beria I, Geroni C, Capolongo L, D'Incalci M, Broggini M (1998) Sequence specific alkylation of novel tallimustine derivatives. _Anticancer Drug Design_
13: 193–205 CAS Google Scholar * Martin DG, Biles C, Gerpheide SA, Hanka LJ, Krueger WC, McGovren JP, Mizsak SA, Neil GL, Stewart JC, Visser J (1981) CC-1065 (NSC 298223), a potent new
antitumor agent improved production and isolation, characterization and antitumor activity. _J Antibiot Tokyo_ 34: 1119–1125 Article CAS PubMed Google Scholar * Modrich P (1991)
Mechanisms and biological effects of DNA mismatch repair. _Ann Rev Genet_ 25: 229–253 Article CAS PubMed Google Scholar * Mosmann T (1983) Rapid colorimetric assay for cellular growth
and survival: application to proliferation and cytotoxicity assays. _J Immunol Methods_ 65: 55–63 Article CAS PubMed Google Scholar * Peltomaki P (2001) DNA mismatch repair and cancer.
_Mutat Res_ 488: 77–85 Article CAS PubMed Google Scholar * Ponti M, Forrow SM, Souhami RL, D'Incalci M, Hartley JA (1991) Measurement of the sequence specificity of covalent DNA
modification by antineoplastic agents using Taq DNA polymerase. _Nucleic Acids Res_ 19: 2929–2933 Article CAS PubMed PubMed Central Google Scholar * Reynolds VL, Molineux IJ, Kaplan DJ,
Swenson DH, Hurley LH (1985) Reaction of the antitumor antibiotic CC-1065 with DNA. Location of the site of thermally induced strand breakage and analysis of DNA sequence specificity.
_Biochemistry_ 24: 6228–6237 Article CAS PubMed Google Scholar * Schwarz VA, Hornung R, Fedier A, Walt H, Haller U, Fink D (2002) Photodynamic therapy of DNA mismatch repair-deficient
and -proficient tumour cells. _Br J Cancer_ 86: 1130–1135 Article CAS PubMed PubMed Central Google Scholar * Smith GC, Cary RB, Lakin ND, Hann BC, Teo SH, Chen DJ, Jackson SP (1999)
Purification and DNA binding properties of the ataxia-telangiectasia gene product ATM. _Proc Natl Acad Sci USA_ 96: 11134–11139 Article CAS PubMed PubMed Central Google Scholar * Sun D,
Hurley LH (1992) Effect of the (+)-CC1065-(N3-adenine) DNA adduct on _in vitro_ DNA synthesis mediated by _Escherichia coli_ DNA polymerase. _Biochemistry_ 31: 2822–2829 Article CAS
PubMed Google Scholar * Umar A, Koi M, Risinger JI, Glaab WE, Tindall KR, Kolodner RD, Boland CR, Barrett JC, Kunkel TA (1997) Correction of hypermutability,
_N_-methyl-_N_′-nitro-_N_-nitrosoguanidine resistance, and defective DNA mismatch repair by introducing chromosome 2 into human tumor cells with mutations in MSH2 and MSH6. _Cancer Res_ 57:
3949–3955 CAS PubMed Google Scholar * Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J (2000) BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair
of aberrant DNA structures. _Genes Dev_ 14: 927–939 CAS PubMed PubMed Central Google Scholar * Westphal CH, Schmaltz C, Rowan S, Elson A, Fisher DE, Leder P (1997) Genetic interactions
between ATM and p53 influence cellular proliferation and irradiation-induced cell cycle checkpoints. _Cancer Res_ 57: 1664–1667 CAS PubMed Google Scholar * Zeng M, Narayanan L, Xu XS,
Prolla TA, Liskay MR, Glazer PM (2000) Ionizing radiation-induced apoptosis via separate pms2- and p53-dependent pathways. _Cancer Res_ 60: 4889–4893 CAS PubMed Google Scholar Download
references ACKNOWLEDGEMENTS We are grateful to Dres M Koi (Laboratory of Molecular Carcinogenesis, National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA), P
Glazer (Yale University School of Medicine, New Haven, CT, USA), E Goodwin (Bioscience Division, Los Alamos National Laboratory, Los Alamos, NM, USA), and P Leder (Department of Genetics and
Howard Hughes Medical Institute, Harvard Medical School, Boston, MA, USA) for generously providing the cell lines. This work has been sponsored by the Cancer League of Canton Zurich and by
an unrestricted grant from Pharmacia AG Switzerland (Dübendorf, Switzerland). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Obstetrics and Gynaecology, Division of Gynaecology,
University Hospital of Zurich, CH-8091, Switzerland A Fedier, U Haller & D Fink * Pharmacia Italy, Oncology, Nerviano, Italy C Fowst, J Tursi & C Geroni * Istituto di Ricerche
Farmacologiche Mario Negri, Milano, Italy S Marchini Authors * A Fedier View author publications You can also search for this author inPubMed Google Scholar * C Fowst View author
publications You can also search for this author inPubMed Google Scholar * J Tursi View author publications You can also search for this author inPubMed Google Scholar * C Geroni View author
publications You can also search for this author inPubMed Google Scholar * U Haller View author publications You can also search for this author inPubMed Google Scholar * S Marchini View
author publications You can also search for this author inPubMed Google Scholar * D Fink View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING
AUTHOR Correspondence to D Fink. RIGHTS AND PERMISSIONS From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share
Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/ Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Fedier, A.,
Fowst, C., Tursi, J. _et al._ Brostallicin (PNU-166196) – a new DNA minor groove binder that retains sensitivity in DNA mismatch repair-deficient tumour cells. _Br J Cancer_ 89, 1559–1565
(2003). https://doi.org/10.1038/sj.bjc.6601316 Download citation * Received: 03 April 2003 * Revised: 08 August 2003 * Accepted: 21 August 2003 * Published: 14 October 2003 * Issue Date: 20
October 2003 * DOI: https://doi.org/10.1038/sj.bjc.6601316 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a
shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative KEYWORDS * brostallicin * DNA mismatch
repair * drug sensitivity * DNA minor groove binder