The p75 receptor mediates axon growth inhibition through an association with PIR-B
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

The Nogo receptor and paired immunoglobulin-like receptor B (PIR-B) are receptors for three myelin-derived axon-growth inhibitors, including myelin-associated glycoprotein (MAG). In this
study, we report that the p75 receptor is required for the signal transduction of PIR-B, which interacted with p75 upon ligand binding. In addition, p75 was required for activation of Src
homology 2-containing protein tyrosine phosphatase (SHP), which is induced by MAG binding to PIR-B. Mice carrying a mutation in the p75 gene showed promotion of axonal regeneration after
optic nerve injury. Thus, our results indicate that p75 has a critical role in axon growth inhibition in specific neuronal tracts.
Axons in the adult mammalian central nervous system (CNS) have very limited ability to regenerate after injury.1 Three myelin-associated, structurally distinct proteins—MAG, Nogo, and
oligodendrocyte myelin glycoprotein (OMgp)—are considered to be involved in this inhibition of axonal regeneration. These three proteins all bind to the Nogo receptor (NgR) and PIR-B. For
signal transduction mediated by NgR, which has no intracellular domain, p75 associates with NgR to transduce the signal inside the cells,1 acting as a displacement factor that releases RhoA
from Rho GDP-dissociation inhibitor.2 This activation of RhoA has a key role in inducing inhibition of axon growth. In contrast, PIR-B is known to be a receptor for MHC class I and functions
as an inhibitory receptor in B cells and myeloid cells.3 The immunoreceptor tyrosine-based inhibitory motifs (ITIMs) of PIR-B recruit SHP-1 and SHP-2, which, in turn, modulate immune
responses. In neurons, PIR-B associates with and inactivates tropomyosin-receptor kinase (Trk) neurotrophin receptors through SHP-1/2.4 Although this reaction is responsible for axon growth
inhibition, the precise mechanism of signal transduction mediated by PIR-B remained to be determined.
p75 is a receptor for neurotrophins and modulates the function of Trk receptors.5 In some cases, p75 opposes the function of Trk receptors and induces cell death; in other cases, it promotes
the function of Trk receptors.6 These findings prompted us to hypothesize that p75 is involved in the PIR-B-mediated inhibition of Trk receptors.
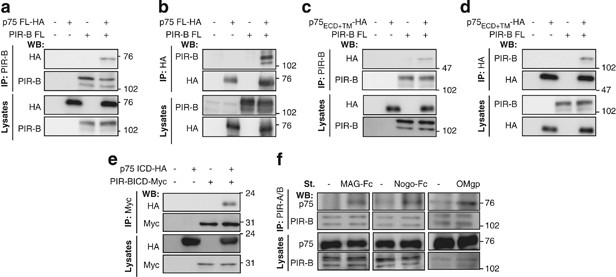
We first assessed the possible involvement of p75 in the PIR-B signal transduction pathway by performing co-immunoprecipitation experiments in COS-7 cells transfected with plasmids encoding
carboxy-terminal hemagglutinin (HA)-tagged full-length p75 (p75 FL-HA) and/or full-length PIR-B (PIR-B FL). Immunoprecipitation was performed using anti-PIR-B or anti-HA antibodies. Of the
PIR-B immunoprecipitates, p75 FL-HA was only detected in the p75 FL-HA/PIR-B FL co-transfected cells (Figure 1a). A complementary experiment revealed that PIR-B was also
co-immunoprecipitated when an anti-HA antibody was used to immunoprecipitate p75-HA (Figure 1b), providing evidence that p75 associates with PIR-B in transfected COS-7 cells.
p75 interacts with PIR-B. (a and b) Co-immunoprecipitation assays to assess the interaction of PIR-B with p75 in COS-7 cells transiently transfected with the indicated plasmids. Lysates were
immunoprecipitated with anti-PIR-B (a) or anti-HA (b) antibodies, and western blotting was carried out with the indicated antibodies. Co-immunoprecipitation assays were also used to examine
interactions between the following exogenously expressed proteins in transfected COS-7 cells: (c and d) full-length PIR-B with HA-tagged p75 ECD + TM, and (e) HA-tagged p75 ICD with
Myc-tagged PIR-B ICD. (f) Co-immunoprecipitation assay demonstrating the interaction of endogenous PIR-B with p75 in unstimulated CGNs or CGNs stimulated with MAG-Fc (25 μg/ml), Nogo-Fc (200
nM), or OMgp (2 μg/ml) for 30 min. Cell lysates were immunoprecipitated with an anti-PIR-A/B antibody, and western blots were performed with an anti-p75 antibody. (a–f) are representative
images from three to four independent experiments. IP, immunoprecipitation
Co-immunoprecipitation experiments were then used to identify the molecular determinants of the p75–PIR-B interaction. COS-7 cells were transfected with the PIR-B FL construct, and/or with a
construct containing the HA-tagged transmembrane domain and extracellular domain of p75 (p75 ECD+TM-HA). Exogenous p75 ECD+TM-HA was found to co-immunoprecipitate with PIR-B FL when
immunoprecipitation was performed with both anti-PIR-B antibodies (Figure 1c) and anti-HA antibodies (Figure 1d). COS-7 cells were then transfected with constructs coding for an HA-tagged
p75 intracellular domain (p75-ICD-HA) and/or Myc-tagged PIR-B ICD (PIR-B ICD-Myc). The interactions between these ectopically expressed proteins were then examined (Figure 1e), and it was
found that the exogenous p75 and PIR-B proteins interact via the intracellular domains, as well as the extracellular domains.
The ligand-dependent interaction of endogenous p75 and PIR-B proteins was also examined in postnatal (day 7) cerebellar granule neurons (CGNs). The CGNs were treated with or without MAG-Fc,
Nogo-Fc, or OMgp for 15 min, and the cell lysates immunoprecipitated with PIR-A/B antibody. It was found that the interaction of p75 with PIR-B was enhanced following ligand stimulation
(Figure 1f), suggesting that ligand binding to PIR-B promoted the interaction of endogenous PIR-B with p75 in the CGNs.
As PIR-B associates with Trk receptors to mediate axon growth inhibition,4 we hypothesized that p75 may also be involved in this signaling complex. The observation that p75 is co-localized
with PIR-B and TrkB in immunostained CGNs supports this theory (Figure 2a). In order to further test whether this is the case, we again performed co-immunoprecipitation. To this end, COS-7
cells were co-transfected with constructs for HA-tagged full-length TrkB (TrkB FL-HA), PIR-B FL, and p75 FL-Myc, and the cell lysates were immunoprecipitated with anti-HA, anti-Myc, or
anti-PIR-A/B antibodies. Each co-immunoprecipitation experiment detected interactions between TrkB FL-HA, p75 FL-Myc, and PIR-B (Figure 2b). Consistent results were obtained when TrkA was
investigated (Figure 2c). The ligand dependency of these interactions was then assessed in CGNs expressing all of these proteins endogenously. The CGNs were treated with or without MAG-Fc or
OMgp for 15 min and immunoprecipitated with anti-TrkB, anti-p75, or anti-PIR-B antibodies. TrkB, p75, and PIR-B were again found to interact following both MAG-Fc treatment (Figure 2d,
left) and OMgp treatment (Figure 2d, right), supporting the hypothesis that p75 interacts with TrkB and PIR-B upon ligand binding with PIR-B.
Interactions between PIR-B, TrkB, and p75 in CGNs. (a) CGNs immunostained for p75 (green), PIR-B (red), and TrkB (blue). Scale Bar: 10 μm. (b and c) Co-immunoprecipitation of the Trk
receptors, p75, and PIR-B in COS-7 cells co-expressing combinations of PIR-B FL, and/or p75 FL-Myc with (b) TrkB FL-HA or (c) TrkA FL-HA. Lysates were immunoprecipitated with the indicated
antibodies, followed by western blotting. IP, immunoprecipitation; WB, western blot. (d) Co-immunoprecipitation assay of CGNs treated with or without MAG-Fc or OMgp for 15 min. Lysates were
immunoprecipitated with the indicated antibodies, and western blotting was carried out. (e) Co-immunoprecipitation of TrkB and PIR-B from CGNs obtained from WT and p75-deficient mice (p75
KO). CGNs were treated with or without MAG-Fc for 15 min. (f) Co-immunoprecipitation of exogenously expressed TrkB-HA and p75 FL-Myc with PIR-B FL from COS-7 cells. The TrkB-HA expression
vectors used were WT TrkB, or TrkB with mutations at Tyr-515 (Tyr-515F), Tyr-705/706 (Tyr-705/706F), or both (Tyr-515/705/706F). Lysates were immunoprecipitated with anti-PIR-A/B antibodies,
followed by western blotting with the indicated antibodies
We subsequently assessed the requirement of p75 for the association between TrkB and PIR-B by examining the ligand-dependent interaction between TrkB and PIR-B in CGNs in mice carrying a
mutation in the p75 gene.7 The ligand-dependent interaction was found to be the same in wild-type (WT) mice and in mice carrying a mutation in the p75 gene (Figure 2e). This is consistent
with the results obtained from transfected COS-7 cells (Figure 2b), suggesting that p75 is involved in the PIR-B/Trk receptor complex, but it may not be necessary for the interaction between
PIR-B and TrkB.
We also examined whether the kinase activity of TrkB is required for its association with PIR-B. It has previously been reported that Tyr-705/706 of TrkB is phosphorylated upon ligand
binding, and that phosphorylation of the Tyr-515 of TrkB is required for interactions with adapter proteins.5 In our experiments, although PIR-B was co-immunoprecipitated with wild-type
TrkB, the level of co-immunoprecipitation with TrkB mutated at Tyr-515 or Tyr-705/706 was significantly reduced (Figure 2f), providing evidence that the kinase activity of TrkB is required
for the interaction between TrkB and PIR-B.
We next examined whether p75 is required for the effects mediated by MAG in neurons. Although MAG inhibits neurite growth in vitro, this effect is dependent on MAG binding to both NgR and
PIR-B. To separate the PIR-B signal from the NgR signal, we focused on the recent finding that MAG stimulation induces dephosphorylation of Trk receptors, which is required for
PIR-B-mediated axon growth inhibition.4 We therefore transfected PIR-B, HA-TrkB, and/or p75-Myc constructs into COS-7 cells, which, as assessed by immunoblot analysis, do not express
detectable levels of endogenous PIR-B, TrkB, or p75 (data not shown). These cells were treated with MAG-Fc for 30 min, and the TrkB receptors were immunoprecipitated. Their phosphorylation
status was then examined using western blotting for phosphotyrosine. It was found that MAG-Fc induced TrkB dephosphorylation in COS-7 cells co-expressing combinations of PIR-B, HA-TrkB, and
p75, but not in cells that did not express p75 (Figure 3a). This requirement for p75 in MAG-Fc functions was confirmed using dissociated retinal neurons isolated from mice carrying a
mutation in the p75 gene. These experiments showed that although TrkB is tyrosine dephosphorylated in response to MAG-Fc treatment in WT-dissociated retinal neurons, there was no change in
TrkB phosphorylation in the neurons isolated from mice bearing the p75 mutation (Figure 3b). Thus, p75 is required for MAG-induced tyrosine dephosphorylation of TrkB receptors. Furthermore,
as COS-7 cells co-expressing combinations of PIR-B, HA-TrkB, and the p75 extracellular domain did not exhibit TrkB dephosphorylation upon MAG stimulation, it suggests that the ICD of p75 is
required for MAG-induced tyrosine dephosphorylation of TrkB (ECD; Figure 3c).
p75 is required for MAG-induced dephosphorylation of TrkB and SHP activation. (a) Western blots examining the level of TrkB tyrosine phosphorylation in transfected COS-7 cells. The cells
were transfected with the indicated constructs and treated with or without MAG-Fc (25 μg/ml) for 30 min in the presence of 100 ng/ml BDNF for 5 min. Anti-Trk antibodies were used to
immunoprecipitate Trk, and the level of Trk phosphorylation was determined by western blotting with anti-phospho-Tyr antibodies. (b–d) Western blots showing the level of TrkB tyrosine
phosphorylation in (b) dissociated retinal neurons from WT and p75 KO mice, (c) transfected COS-7 cells co-expressing TrkB FL-HA and PIR-B FL in combination with p75 FL-Myc or p75 ECD-Myc,
and (d) COS-7 cells transfected with or without a construct expressing catalytically inactive SHP-2 (SHP-2 C459S). (e) Western blots showing the expression of the indicated proteins in WT
and p75 KO CGNs. (f) Western blots for detecting the levels of phospho-SHP-2 (pTyr-580) and total SHP-2 in CGNs following treatment with MAG-Fc for the indicated periods. FGF (50 ng/ml) was
used as a positive control. The relative intensity for the phosphorylated SHP-2 signal is indicated (mean: n=3). (g) Western blots showing phospho-SHP-2 (pTyr-580) and total SHP-2 levels in
WT and p75 CGNs. The graph shows relative phospho-SHP-2 levels. (h) Relative SHP-2 PTP activity as measured by PTP assays in CGNs from WT (left) and p75 KO (right) mice. Cells were treated
with MAG-Fc for 20 min. *P