Interaction between autophagy and senescence is required for dihydroartemisinin to alleviate liver fibrosis
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Autophagy and cellular senescence are stress responses essential for homeostasis. Therefore, they may represent new pharmacologic targets for drug development to treat diseases. In
this study, we sought to evaluate the effect of dihydroartemisinin (DHA) on senescence of activated hepatic stellate cells (HSCs), and to further elucidate the underlying mechanisms. We
found that DHA treatment induced the accumulation of senescent activated HSCs in rat fibrotic liver, and promoted the expression of senescence markers p53, p16, p21 and Hmga1 in cell model.
Importantly, our study identified the transcription factor GATA6 as an upstream molecule in the facilitation of DHA-induced HSC senescence. GATA6 accumulation promoted DHA-induced p53 and
p16 upregulation, and contributed to HSC senescence. By contrast, siRNA-mediated knockdown of GATA6 dramatically abolished DHA-induced upregulation of p53 and p16, and in turn inhibited HSC
senescence. Interestingly, DHA also appeared to increase autophagosome generation and autophagic flux in activated HSCs, which was underlying mechanism for DHA-induced GATA6 accumulation.
Autophagy depletion impaired GATA6 accumulation, while autophagy induction showed a synergistic effect with DHA. Attractively, p62 was found to act as a negative regulator of GATA6
accumulation. Treatment of cultured HSCs with various autophagy inhibitors, led to an inhibition of DHA-induced p62 degradation, and in turn, prevented DHA-induced GATA6 accumulation and HSC
senescence. Overall, these results provide novel implications to reveal the molecular mechanism of DHA-induced senescence, by which points to the possibility of using DHA based
proautophagic drugs for the treatment of liver fibrosis. SIMILAR CONTENT BEING VIEWED BY OTHERS IMPLICATION OF AUTOPHAGY IN THE ANTIFIBROGENIC EFFECT OF RILPIVIRINE: WHEN MORE IS LESS
Article Open access 20 April 2022 GINSENOSIDE RG3 PROMOTES REGRESSION FROM HEPATIC FIBROSIS THROUGH REDUCING INFLAMMATION-MEDIATED AUTOPHAGY SIGNALING PATHWAY Article Open access 12 June
2020 CARFILZOMIB SHOWS THERAPEUTIC POTENTIAL FOR REDUCTION OF LIVER FIBROSIS BY TARGETING HEPATIC STELLATE CELL ACTIVATION Article Open access 20 August 2024 MAIN Liver fibrosis is a
reversible wound-healing response following liver injury, and its end-stage cirrhosis is responsible for high morbidity and mortality worldwide.1, 2, 3 Liver transplantation is the only
treatment available for patients with advanced stages of liver fibrosis.4, 5, 6 Therefore, new therapeutic agents and strategies are needed for the management of this disease.7, 8
Dihydroartemisinin (DHA), a natural and safe anti-malarial agent, exhibits an ample array of pharmacological activities such as anti-tumor,9 anti-bacterial10 and anti-schistosomiasis
properties.11 We previously reported that DHA treatment improved the inflammatory microenvironment of liver fibrosis _in vivo_,12 and inhibited activation and contraction of hepatic stellate
cells (HSCs) _in vitro._13, 14, 15, 16 In the current study, we aimed to evaluate the effect of DHA on HSC senescence and to further elucidate the underlying mechanisms. Cellular senescence
is a terminal arrest of proliferation triggered by various cellular stresses including dysfunctional telomeres,17 DNA damage18 and oncogenic mutations.19 Cellular senescence not only
prevents the proliferation of damaged cells, thereby preventing tumorigenesis, but also affects the microenvironment through the secretion of pro-inflammatory cytokines, chemokines, and
proteases, a feature termed the senescence-associated secretory phenotype (SASP).20 The mechanisms underlying induction and maintenance of cell senescence remain entirely elusive.21, 22, 23
Previous studies21, 22 have reported that p53 can lead to cell cycle arrest, DNA repair and apoptosis predominantly when it becomes transcriptionally active in response to DNA damage,
oncogene activation and hypoxia. Retinoblastoma 1 (pRb) inactivation mediated by p16 is also known to ensure durable cell cycle arrest, but is unlikely to be regulated by a canonical DNA
damage response.23 Attractively, it is interesting to explore the mechanism underlying the induction and maintenance of cell senescence in liver fibrosis. Interestingly, several lines of
evidence indicate a genetic relationship between autophagy and senescence.24, 25 However, whether autophagy acts positively or negatively on senescence is still subject to debate.25 Through
a specialized compartment known as the TOR-autophagy spatial coupling compartment (TASCC), autophagy generates a high flux of recycled amino acids, which are subsequently used by mTORC1 for
supporting the massive synthesis of the SASP factors and facilitating senescence.25 In contrast, increased levels of reactive oxygen species upon autophagy inhibition partially contribute to
cellular senescence.25 We previously reported that DHA treatment stimulated autophagy activation via a ROS-JNK1/2-dependent mechanism in liver fibrosis.12 Attractively, whether autophagy
activation contributes to DHA-induced HSC senescence is worth to further study. In the present study, we evaluated the effect of DHA on HSC senescence, and to further elucidate the
underlying mechanisms. We found that DHA could induce senescence of activated HSCs to alleviate liver fibrosis via autophagy-dependent GATA6 accumulation. The results of the present study
provide important information concerning the molecular mechanisms that underlie the antifibrotic activities of DHA, which is essential for investigating its potential for clinical
application. RESULTS DHA INDUCES THE ACCUMULATION OF SENESCENT ACTIVATED HSCS IN RAT FIBROTIC LIVER Our previous data12, 13, 14, 15, 16 and the present results (Supplementary Figures 1A–C)
have sufficiently demonstrated that DHA protected the liver against CCl4-induced injury and suppressed hepatic fibrogenesis in the rat model. To investigate the mechanisms underlying the
protective effects of DHA, we proposed that DHA might induce senescence of activated HSCs to limit liver fibrosis. To identify senescent cells _in situ_, we stained liver sections from DHA
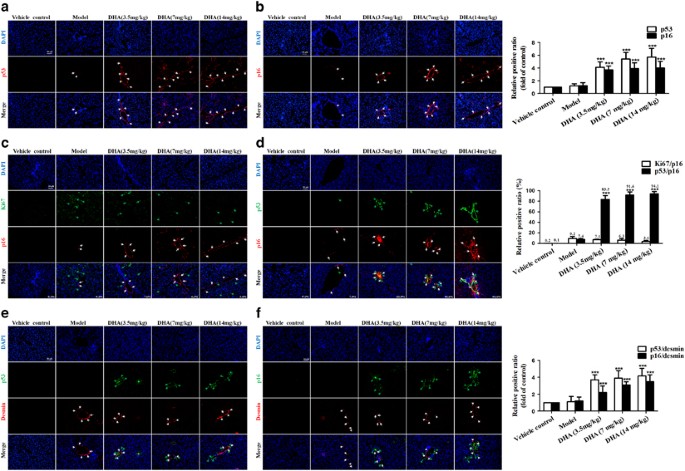
and vehicle-treated rat for a panel of senescence-associated markers, including SA-_β_-gal, p53 and p21. Results from immunofluorescence staining showed that cells staining positive for each
senescence-associated markers accumulated in fibrotic livers, and were invariably located along the fibrotic scar by treatment with DHA in a dose-dependent manner (Figures 1a and b;
Supplementary Figure 1D). Interestingly, we also found that these cells typically expressed multiple senescence markers and were not proliferating. As shown in Figures 1c and d, of the
p16-positive cells identified in DHA-treated livers, more than 80% were positive for p53 staining, whereas less than 9% co-expressed the proliferation-association marker Ki67. Although
hepatocytes represent the most abundant cell type in the liver, the location of senescent cells along the fibrotic scar in rat livers raised the possibility that these cells were derived
from activated HSCs, which initially proliferate following liver damage and are responsible for much of the extracellular matrix production in fibrosis.4, 5, 6 In order to verify this
hypothesis, the cells in DHA- and vehicle-treated liver sections were not only stained positive for the senescence-associated markers p53 and p16, but also were positive for the HSC marker
desmin. As expected, cells expressing the senescence markers p53 and p21 co-localized with those expressing desmin (Figures 1e and f). Overall, these results indicate that DHA induces the
accumulation of senescent activated HSCs in rat fibrotic liver. DHA PROMOTES ACTIVATED HSC SENESCENCE _IN VITRO_ Previous studies have confirmed that HSC activation _in vivo_, as a result of
different liver injuries, could be mimicked _in vitro_ by plating freshly isolated HSCs exposed to platelet derived growth factor-BB (PDGF-BB) on plastic tissue culture dishes.12, 13, 14,
15, 16, 26 Therefore, the freshly quiescent HSCs were isolated from Sprague-Dawley rats as described,27 and then were treated with 5, 10 and 20 ng/ml PDGF-BB. In agreement with previous
findings,12, 13, 14, 15, 16 HSC activation markers like _α_-SMA (acta-2), Fibronectin, Procollagen 1_α_1 (procol1_α_1), TNF-_α_, and TGF-_β_ were significantly upregulated showing that HSCs
undergo an activation process _in vitro_ as well (Supplementary Figures 2A–C). Subsequently, we used cultured HSCs to test whether DHA treatment could promote activated HSC senescence _in
vitro_. Immunofluorescent assay showed that DHA and Etoposide (as a positive control)28 treatment significantly increased the expression of senescence markers p53, p16, p21, and Hmga1 in
cell model (Figure 2a). Besides, we found that DHA treatment increased the number of SA-_β_-Gal-positive HSCs (Figure 2b). Western blot and Real-time PCR analysis of senescence-associated
proteins also consistently showed that DHA treatment upregulated the expression of p53, p16, p21 and Hmga1 in activated HSCs (Figures 2c and d). Additional experiments were performed to
verify the role of telomerase activity in DHA-induced HSC senescence. We found that the telomerase activity was decreased in DHA-treated HSCs (Supplementary Figure 2D). A well-known feature
of cellular senescence is cell cycle arrest, which largely accounts for the growth inhibition in senescent cells.20 Next, we examined the cell cycle distribution by a flow cytometer. As
shown in Supplementary Figures 2E and F, HSCs treated with DHA or Etoposide showed significantly higher proportions of G2/M cells and lower proportions of S cells compared with untreated
HSCs. Cell cycle is influenced by multiple cyclins and cyclin-dependent kinases (CDKs).29 Real-time PCR analyses indicated that DHA treatment downregulated the expression of cyclin D1,
cyclin E1 and CDK4 in activated HSCs (Supplementary Figure 2G). Taken together, these results show that DHA promotes activated HSC senescence _in vitro_. THE ACCUMULATION OF GATA6 IS
REQUIRED FOR DHA TO INDUCE ACTIVATED HSC SENESCENCE _IN VITRO_ Cellular senescence is a terminal stress-activated program mainly controlled by the p53 and p16INK4a tumor suppressor
proteins.22, 23 However, in contrast to the downstream functionality of p53 and p16, its upstream control is a relatively unexplored area.30 In the current study, we hypothesized that GATA6
could have a pivotal role in DHA-induced upregulation of p53 and p16 in activated HSCs. To test this hypothesis, the status of this GATA6 protein was evaluated following the DHA treatment.
As shown in Figure 3a, DHA treatment obviously increased the level of GATA6 in a time- and dose-dependent manner. In order to further detect the role of GATA6 accumulation in the DHA-induced
senescence, activated HSCs were pre-treated with GATA6 siRNA or GATA6 plasmid, followed by DHA treatment (Figure 3b). As expected, the results from SA-_β_-Gal staining showed that
pretreatment with GATA6 siRNA significantly abrogated DHA-induced increase of SA-_β_-Gal-positive HSCs, whereas GATA6 plasmid showed a synergistic effect with DHA (Figures 3e and h).
Besides, in order to investigate the effect of GATA6 accumulation on DHA-induced p53 and p16 upregulation, the expression of p53, p16, and their downstream effectors were detected by western
blot and Real-time PCR analysis. The results revealed that GATA6 plasmid, mimicking DHA, promoted the expression of p53, p21, Hmga1 and p16, while GATA6 siRNA dramatically suppressed the
ability of DHA and GATA6 plasmid in inducing cellular senescence (Figures 3c, d, f and g). Furthermore, immunofluorescent assay also indicated that DHA as well as GATA6 plasmid significantly
increased the abundance of proteins involved in senescence (Supplementary Figures 3B and D). However, the pretreatment of cells with GATA6 siRNA dramatically eliminated the promoting
effects of DHA on the expression of p53 and p16 in activated HSCs (Supplementary Figures 3A and C). Attractively, accumulating evidence suggests that mitogen activated protein kinases
(MAPKs) have important roles in the activation of p53 and p16.31, 32, 33 Thus, it was assumed that GATA6 accumulation contributed to DHA-induced upregulation of p53 and p16 via a
MAPK-dependent mechanism. To test this assumption, the phosphorylation status of these MAPK proteins was evaluated following the GATA6 siRNA or GATA6 plasmid treatment. The results showed
that GATA6 plasmid obviously increased the level of phosphorylated JNK1/2, but did not significantly affect the level of phospho-ERK1/2 and phospho-p38 (Supplementary Figure 3E), suggesting
the involvement of JNK1/2 in GATA6-induced upregulation of p53 and p16. In order to further determine the association between GATA6 accumulation and p53 or p16 upregulation, selective JNK1/2
inhibitor (SP600125) was used to inhibit the activity of JNK1/2. Interestingly, we found that pretreatment with SP600125 abolished GATA6 plasmid or DHA-induced p53 and p16 upregulation
(Supplementary Figures 3F and G), demonstrating that JNK1/2 could mediate p53 and p16 upregulation induced by GATA6 accumulation. Collectively, these data demonstrate that the accumulation
of GATA6 is required for DHA to induce HSC senescence _in vitro_. DHA INDUCES HSC SENESCENCE VIA A GATA6-DEPENDENT MECHANISM _IN VIVO_ We further examined whether the disruption of GATA6
accumulation could affect DHA-induced upregulation of p53 and p16 _in vivo_. Seventy mice were randomly divided into seven groups of ten animals each with comparable mean bodyweight. Mice of
seven groups were administrated with vehicle control, CCl4, CCl4+Ad.Fc, CCl4 +DHA, CCl4+Ad.Fc+DHA, CCl4+Ad.shGATA6 or CCl4+Ad.shGATA6+DHA, respectively, throughout the 8-week period of CCl4
treatment. First and foremost we evaluated the effect of interrupting GATA6 on liver injury _in vivo_. Gross examination showed that morphological changes pathologically occurred in the
mouse liver exposed to CCl4 compared with the normal liver, but DHA treatment improved the pathological changes in livers (Figure 4a). Interestingly, the improvement of DHA on liver injury
was remarkably abrogated by Ad.shGATA6 (Figure 4a). Besides, liver fibrosis was also demonstrated by histological analyses. Hematoxylin and eosin (H&E), Masson and picro-Sirius red
staining showed that intraperitoneal injection of DHA daily for 4 weeks significantly improved histopathological feature of liver fibrosis characterized by decreased collagen deposition,
whereas livers derived from mice treated with DHA plus Ad.shGATA6 exhibited more severe liver fibrosis compared with the mice treated with DHA alone (Figure 4a). Next, primary HSCs were
isolated for detection of cell senescence markers. The Real-time PCR analysis showed that Ad.shGATA6 significantly reduced the GATA6 level of activated HSCs (Figure 4b). Then, western blot
and Real-time PCR analysis demonstrated that interference of GATA6 significantly inhibited the expression of p53, p21 and p16, suggesting that the effect of DHA was at least partially
reversed (Figures 4c and e). Besides, Ad.shGATA6 treatment not only decreased the number of SA-_β_-Gal-positive cells, but also markedly eliminated the regulatory effects of DHA on cell
senescence (Figure 4d). More importantly, liver tissues were co-stained with the senescence markers p53 or p16 and HSC activation marker desmin. Results from immunofluorescence staining
showed that DHA induced the accumulation of senescent activated HSCs in fibrotic liver, whereas Ad.shGATA6 treatment impaired the induction of DHA on activated HSC senescence (Figure 4f).
Altogether, these data suggest that GATA6 accumulation is involved in DHA-induced HSC senescence _in vivo_. THE ACTIVATION OF AUTOPHAGY IS ASSOCIATED WITH DHA-INDUCED GATA6 ACCUMULATION AND
HSC SENESCENCE Protein accumulation is controlled by two major pathways in eukaryotic cells: the ubiquitin-proteasome34 and autophagy-lysosome pathways.35 Interestingly, Kang _et al._36
showed that inhibition of the proteasome by MG-132, a proteasome inhibitor, had no effect on GATA4 abundance, whereas GATA4 protein was stabilized in cells treated with distinct lysosomal
inhibitors known to block autophagy. In the present study, we assumed that autophagy-lysosome pathways could have a pivotal role in DHA-induced GATA6 accumulation. To evaluate this
assumption, activated HSCs were treated with various concentrations of DHA for 24 h or with 20 _μ_M of DHA for different hours. Results from western blot analysis showed that DHA induced the
generation of autophagosome in a dose- and time-dependent manner (Figure 5a). Besides, seven important autophagy related genes were detected by western blot and Real-time PCR analysis in
DHA- or vehicle-treated cells. The results revealed that DHA treatment increased the level of many indicators of the autophagosome (Figure 5b). Furthermore, immunofluorescence of
Atg6/Beclin1 and endogenous LC3-II also proved the facilitating roles of DHA on autophagosome (Supplementary Figures 4C and D). Numerous studies have shown a crucial role for mTOR signaling
pathway in autophagosome generation.37, 38 Therefore, we evaluated whether DHA treatment affect the expression of p-ULK1, ULK1, p-mTOR and mTOR. Western blot analysis revealed that
DHA-induced autophagosome generation was associated with an increase in p-ULK1 activity and a decrease in p-mTOR activity (Supplementary Figure 4A). Next, we further assessed the effect of
DHA on autophagic flux in activated HSCs. Firstly, tandem fluorescence RFP-GFP-LC3 (tf-LC3) staining was used to demonstrate the autophagic flux. It has been documented that, in
autophagosomes, the combination of both RFP and GFP in the triple fusion yields yellow fluorescence, whereas autolysosomal delivery results in red.39 As expected, we observed that both
autophagosome and autolysosome formation were increased in DHA-treated HSCs (Figure 5c). Secondly, the long-lived protein degradation was detected to indicate autophagic flux because it was
substrate for autophagy, and the rate was a key functional index of autophagic flux.39 The longevity protein degradation rate reflected that DHA time-dependently increased autophagic flux
(Figure 5d). Thirdly, we observed an increase in LC3-II level in cells which cultured with DHA for 24 h followed by chloroquine (CQ) treatment compared with cells which were treated with DHA
alone, suggesting that autophagic flux is increased in DHA-treated HSCs (Supplementary Figure 4B). Lastly, the transmission electron microscopy (TEM) was used to observe autophagy.39 As
expected, we observed the presence of a high level of autophagosomes or lysosomes in DHA-treated HSCs. In contrast, it was difficult to observe autophagosomes or lysosomes in control HSCs
(Figure 5e). Overall, these results support that DHA increases the autophagosome generation and autophagic flux in activated HSCs. DISRUPTION OF AUTOPHAGY IMPAIRS DHA-INDUCED GATA6
ACCUMULATION AND HSC SENESCENCE _IN VITRO_ To determine whether the activation of autophagy by DHA is directly involved in the GATA6 accumulation and HSC senescence _in vitro_, we used Atg5
siRNA to block the autophagosome formation and employed Atg5 plasmid to induce autophagy (Figures 6a and b). Then, SA-_β_-Gal staining was performed to measure its effects on cell
senescence. As shown in Figure 6c, Atg5 plasmid, mimicking DHA, increased the number of SA-_β_-Gal-positive HSCs. Conversely, siRNA-mediated knockdown of Atg5 markedly suppressed the ability
of DHA and Atg5 plasmid in the induction of cell senescence. Furthermore, Real-time PCR analysis indicated that the pretreatment of cells with Atg5 siRNA significantly altered the abundance
of p53 and p16 mRNA induced by DHA treatment (Figure 6d). Besides, the results from immunofluorescence assay showed that DHA as well as Atg5 plasmid treatment significantly increased the
level of cellular GATA6 and p53 compared with untreated cells, whereas the treatment of cells with Atg5 siRNA, which downregulated the expression of cellular GATA6 and p53, dramatically
diminished the effect of DHA or Atg5 plasmid in inducing cell senescence(Figure 6e). Additional experiments showed that DHA and Atg5 plasmid treatment significantly inhibited the telomerase
activity, whereas the pretreatment with Atg5 siRNA abrogated DHA-induced inhibitory effects (Supplementary Figure 5A). Lastly, we examined the effect of Atg5 plasmid or Atg5 siRNA on cell
cycle distribution. As shown in Supplementary Figures 5B–D, the pretreatment with Atg5 plasmid decreased the expression of Cyclin D1, CDK4 and CDK6, while siRNA-mediated knockdown of Atg5
dramatically upregulated their expression and resulted in a pronounced and significant attenuation of DHA-induced inhibitory effects. Collectively, these results support that autophagy
activation mediates DHA-induced GATA6 accumulation and cell senescence in activated HSCs. DEGRADATION OF P62 IS REQUIRED FOR AUTOPHAGY TO MEDIATE DHA-INDUCED GATA6 ACCUMULATION AND HSC
SENESCENCE _IN VITRO_ To further investigate how DHA-induced autophagy promoted GATA6 accumulation, we hypothesized that the degradation of p62 had an important role in DHA-induced GATA6
accumulation and HSC senescence. To test this hypothesis, the status of this p62 protein was evaluated following the DHA treatment. Western blot analysis showed that treatment with DHA for
18 h resulted in a significant inhibitory effect, which was negatively correlated to the GATA6 accumulation of DHA-treated HSCs (Figure 7a). Then, the interaction between p62 and GATA6 was
determined by immunoprecipitation assay. The result revealed that DHA blocked the binding of p62 to GATA6 in a dose-dependent manner (Figure 7b). Interestingly, these data suggest that p62
may be a negative regulator of GATA6 accumulation, but this regulation is suppressed by DHA-induced autophagy activation, thereby stabilizing GATA6. Next, various autophagy inhibitors, 3-MA,
CQ and Bafilomycin A1, were used to induce p62 accumulation for a reverse verification. As shown in Figures 7c and f, treatment with DHA significantly decreased the expression of p62,
whereas pretreatment with 3-MA, CQ, and Bafilomycin A1 completely abrogated DHA-induced p62 degradation. As expected, immunofluorescent staining of GATA6 demonstrated that the stabilization
of p62 induced by autophagy inhibitors 3-MA, CQ and Bafilomycin A1, impaired DHA-induced GATA6 accumulation (Figure 7f). Furthermore, a panel of senescence-associated markers, including
SA-_β_-gal, p53 and p21, were all determined. Unsurprisingly, treatment with 3-MA, CQ, and Bafilomycin A1 completely abrogated DHA-induced p62 degradation, and in turn, decreased the number
of SA-_β_-Gal-positive HSCs (Figure 7d), and p16 mRNA expression (Figure 7e). More importantly, p62 overexpression plasmid also resulted in a pronounced and significant attenuation of
DHA-induced GATA6 accumulation, and then, decreased the number of SA-_β_-Gal-positive HSCs, and p53 or p16 mRNA expression (Supplementary Figures 6A–D). Taken together, these data suggest
that the degradation of p62 is required for autophagy to mediate DHA-induced GATA6 accumulation and HSC senescence _in vitro_. DISCUSSION Cellular senescence acts as a potent mechanism of
tumor suppression.17, 18, 19, 20 However, its functional contribution to non-cancer pathologies has not been fully understood. Attractively, previous studies,22, 40 have discovered the
existence of senescent HSCs during the development of liver fibrosis. Krizhanovsky _et al._ showed that senescent activated HSCs reduced the secretion of ECM components, enhanced immune
surveillance, and facilitated the reversion of fibrosis.40 Kong _et al._ also reported that interleukin-22 induced HSC senescence and restricted liver fibrosis in mice.41 Consistent with
previous studies,40, 41 we showed that the senescence of activated HSCs induced by DHA treatment provide a brake on the fibrogenic response to damage by limiting the expansion of the cell
type responsible for producing the fibrotic scar. To our knowledge, this is the first report that DHA can induce HSC senescence to alleviate liver fibrosis. Importantly, our study identified
the transcription factor GATA6 as an upstream molecule in the facilitation of DHA-induced HSC senescence. The GATA family of transcription factors consists of six proteins (GATA1-6) which
are involved in a variety of physiological and pathological processes.42, 43 Recently, Kang _et al._ reported that GATA4 functioned as a key switch in the senescence regulatory network to
activate the senescence-associated secretory phenotype (SASP).36 In the present study, we found that the accumulation of GATA6 was required for DHA to induce HSC senescence _in vitro_ and
_in vivo_. siRNA-mediated knockdown of GATA6 dramatically abolished DHA-induced upregulation of p53 and p16, and in turn inhibited HSC senescence. Although our data suggested direct
connection between GATA6 and DHA-induced HSC senescence, we could not eliminate other effects that may mediate the protective effect of DHA. Autophagy and cellular senescence are stress
responses essential for homeostasis.24 While recent studies indicate a genetic relationship between autophagy and senescence, whether autophagy acts positively or negatively on senescence is
still subject to debate.24, 25 Garcia-Prat _et al._ reported that autophagy maintains stemness by preventing senescence.44 Conversely, Liu _et al._ revealed that autophagy suppresses
melanoma tumorigenesis by inducing senescence.45 In the current study, we found that activation of autophagy is required for DHA to induce HSC senescence in live animal model and cell model.
Down-regulation of autophagy activity, using Atg5 siRNA, led to an inhibition of DHA-induced HSC senescence, while Atg5 plasmid enhanced the effect of DHA _in vitro_. Attractively, we found
that p62 may be a negative regulator of GATA6 accumulation, but this regulation was suppressed by DHA-induced autophagy activation. Treatment of cultured HSCs with various autophagy
inhibitors or p62 overexpression plasmid, led to an inhibition of DHA-induced p62 degradation, and in turn, prevented DHA-induced GATA6 accumulation and HSC senescence. Although more
experiments are needed to determine the exact role of autophagy in cell senescence, our results indicate a similar function in HSCs in consistent with previous reports.24, 25 Overall, these
results provide the first mechanistic evidence that interaction between autophagy and senescence is required for DHA to alleviate liver fibrosis (Figure 8). Since there still are no
clinically effective anti-fibrosis drugs, understanding the mechanistic basis of action of natural dietary products such as DHA offers further insight into developing drugs for the
prevention and treatment of liver fibrosis. MATERIALS AND METHODS REAGENTS AND ANTIBODIES DHA, colchicine, PDGF-BB, Etoposide, CQ, rapamycin (Rapa), 3-MA, bafilomycin A1, dimethyl sulfoxide
(DMSO), anti-rabbit IgG, and anti-mouse IgG were purchased from Sigma-Aldrich (St. Louis, MO, USA). Dulbecco’s modified essential medium (DMEM), Opti MEM medium, phosphate-buffered saline
(PBS), trypsin-EDTA and fetal bovine serum (FBS) were bought from GIBCO BRL (Grand Island, NY, USA). Primary antibodies against p53, p16, p21, _α_-SMA, Hmga1, LC3-I/II, ULK1, p-ULK1, mTOR,
p-mTOR, Atg3, Atg5-Atg12, Atg6, Atg7, Atg14, p62, _β_-galactosidase and _β_-actin were purchased from Cell Signaling Technology (Danvers, MA, USA). Primary antibody against _α_1(I)
procollagen was purchased from Epitomics (San Francisco, CA, USA). Primary antibodies against Ki67, p62 and GATA6 were purchased from Abcam Technology (Abcam, Cambridge, UK). Atg5 siRNA,
GATA6 siRNA, negative control siRNA, Atg5 plasmid, GATA6 plasmid, negative control vectors and mRFP-GFP-LC3 plasmid were purchased from Hanbio (Shanghai, China). MegaTran 1.0 transfection
reagent was from OriGene (Rockville, MD, USA). ANIMALS AND EXPERIMENTAL DESIGN All experimental procedures were approved by the institutional and local committee on the care and use of
animals of Nanjing University of Chinese Medicine (Nanjing, China), and all animals received humane care according to the National Institutes of Health (USA) guidelines. Male Sprague-Dawley
rats weighing approximately 180–220 g were procured from Nanjing Medical University (Nanjing, China). A mixture of CCl4 (0.1 ml/100g bodyweight) and olive oil (1:1 (w/v)) was used to induce
liver fibrosis in rats. Fifty rats were randomly divided into five groups of ten animals each with comparable mean bodyweight. Rats of Group 1 were served as a vehicle control and
intraperitoneally (i.p.) injected with olive oil. Rats of group 2 were i.p. injected with CCl4. Rats of Groups 3, 4 and 5 were served as treatment groups and i.p. injected by CCl4 and DHA
with 3.5, 7 and 14 mg/kg, respectively. Rats of groups 2–5 were i.p. injected with CCl4 every other day for 8 weeks. DHA was suspended in sterile PBS and given once daily by intraperitoneal
injection during weeks 5–8. At the end of the experiment, rats were sacrificed after anesthetization with an injection of 50 mg/kg pentobarbital. A small portion of the liver was removed for
histopathological and immunohistochemical studies. Male ICR mice (ages 6–8 weeks) were purchased from Nanjing Medical University (Nanjing, China). Seventy mice were randomly divided into
seven groups of ten animals each with comparable mean bodyweight. Mice of seven groups were administrated with Vehicle control, CCl4, CCl4+Ad.Fc (a control adenovirus encoding IgG2 _α_ Fc
fragment), CCl4+DHA (20 mg/kg, once a day), CCl4+Ad.Fc+DHA, CCl4+Ad.shGATA6 (adenovirus encoding mouse GATA6 shRNA for inhibiting GATA6 expression) or CCl4+Ad.shGATA6+DHA, respectively,
throughout the 8-week period of CCl4 treatment. Adenoviruses (2.5 × 107pfu/g, once per 2 weeks) were injected into mice by tail vein. A mixture of carbon tetrachloride (CCl4; 0.5 ml per 100
g bodyweight) and olive oil (1: 9 (v/v)) was used to induce liver fibrosis in mice by i.p. injection. After 8 weeks, liver were fixed in 4% buffered paraformaldehyde for histological
analysis of liver fibrosis and immunostaining analysis or HSCs were isolated for western blot analysis. HISTOLOGICAL ANALYSIS Hematoxylin and eosin, Sirius Red and Masson staining were
performed on 4-_μ_m thick formalin-fixed paraffin-embedded tissue sections. Sirius Red and Masson-stained areas from 10 fields (magnification × 200) from 3 to 6 mice/group were quantified
with Image J. CELL ISOLATION, CELL CULTURE CONDITIONS AND DRUG TREATMENT Primary rat HSCs were isolated from male Sprague-Dawley rats weighing approximately 180–220 g (Nanjing Medical
University, Nanjing, China) as described.27 Isolated HSCs were cultured in DMEM with 10% fetal bovine serum, 1% antibiotics and maintained at 37 °C in a humidified incubator of 5% CO2 and
95% air. Cell morphology was assessed using an inverted microscope with a Leica Qwin System (Leica, Germany). DHA was dissolved in DMSO at a concentration of 10 mM and was stored in a dark
colored bottle at −20 °C. The stock was diluted to required concentration with DMSO when needed. Before the DHA treatment, cells were grown to about 70% confluence, and then exposed to DHA
at different concentrations (0–20 _μ_M) for different period of time (0–24 h). Cells grown in a medium containing an equivalent amount of DMSO without DHA were served as control. PLASMID
TRANSFECTION Atg5 siRNA, GATA6 siRNA, p62 siRNA, negative control siRNA, Atg5 plasmid, GATA6 plasmid, p62 plasmid, negative control vectors and mRFP-GFP-LC3 plasmid were transfected into
HSCs using MegaTran 1.0 transfection reagent according to manufacturer's instructions.12 After 24 h, cells were treated with selenite or PBS as a solution control. The transfection
efficiency was confirmed by western blot analysis. RNA ISOLATION AND REAL-TIME PCR Total RNA was isolated and qPCR performed using the QuantiTect SYBR Green PCR Kit (Qiagen, Valencia, CA,
USA) in accordance to the manufacturer's instructions.12 Actin levels were taken for normalization and fold change was calculated using 2-ddCt. Primer Sequence available on request.
WESTERN BLOT ANALYSIS Cells or tissue samples were lysed using mammalian lysis buffer (Sigma, St. Louis, MO, USA) and immunoblotting was performed as per the manufacturer’s guidelines12
(Bio/Rad, Hercules, CA, USA). Briefly, the protein levels were determined using a BCA assay kit (Pierce, Rockford, IL, USA). Proteins (50 _μ_g/well) were separated by SDS-polyacrylamide gel,
transferred to a PVDF membrane (Millipore, Burlington, MA, USA), blocked with 5% skim milk in Tris-buffered saline containing 0.1% Tween 20. Target proteins were detected by corresponding
primary antibodies, and subsequently by horseradish peroxidase-conjugated secondary antibodies. Protein bands were visualized using chemiluminescence reagent (Millipore). Equivalent loading
was confirmed using an antibody against _β_-actin. Densitometry analysis was performed using Image J software (NIH, Bethesda, MD, USA). IMMUNOPRECIPITATION ASSAY An immunoprecipitation assay
was performed using extracts of the activated HSCs as previously described.46 Briefly, immunoprecipitation was performed using Classic Magnetic IP/Co-IP Kit (Pierce, Carlsbad, CA, USA) to
analyze the interaction between GATA6 and p62 (Abcam, Cambridge, UK). Activated HSCs were washed 3 times in PBS and lysed in IP Lysis Buffer (Abcam) on ice for 5 min. The protein lysate was
collected by centrifugation. Protein A/G Magnetic Beads (25 _μ_l) were incubated with anti-GATA6 antibody (Abcam) for 1 h at room temperature, and then added to the protein lysate and
incubated overnight at 4 °C. The beads were then collected and washed in IP Wash Buffer for 5 times. Proteins were dissolved in Elution Buffer and detected by western blot. LONG-LIVED
PROTEINS DEGRADATION ANALYSIS Primary HSCs were cultured in DMEM (GIBCO BRL) supplemented with 100 units/ml penicillin, 100 _μ_g/ml streptomycin, glutamine, 10% fetal bovine serum (GIBCO
BRL) and labeled with either l-arginine and l-lysine, l-[U-13C6,14N4] arginine and l-[2H4]lysine, or l-[U-13C6,15N4]arginine and l-[U-13C6,15N2]lysine (Cambridge Isotope Laboratories,
Andover, MA, USA; Sigma-Aldrich) (2*15-cm cell culture dishes per condition; ~95% confluent). Cells were washed three times in PBS before they were treated with DMSO or 20 _μ_M DHA following
20 ng/ml PDGF-BB treatment for indicated hours. All treatments were carried out at 37 °C. After drug treatment, the cells were scraped in cell scraping buffer (0.25 M sucrose, 1 mM sodium
ortho-vanadate, 5 mM NaF, 5 mM _β_-glycerophosphate, and protease inhibitor mixture (Complete TM tablets, Roche Diagnostics)) and normalized by cell counting. Mixed cells were centrifuged
for 5 min at 1800 rpm and lysed in 6 M urea and 2 M thiourea, and 2% Benzonase (Merck) was added before samples were concentrated on spin tubes (cutoff, 500 Da). Protein mixtures were
separated by SDS-PAGE (4–12% bis-Tris gra-dient gel, NuPAGE, Invitrogen). Gel lanes were cut into 15 slices, and samples were in-gel digested, and resulting peptide mixtures were
STAGE-tipped. Relative quantification and identification of peptides were analyzed by LC–MS/MS as described previously.39 TRANSMISSION ELECTRON MICROSCOPY Cells were seeded onto 4-chambered
coverglass (Lab-Tek Chambered Coverglass System) (Nalgene/Nunc, Rochester, NY, USA) at a density of 2 × 104 cells/ml (14 000 cells/well). Images were acquired using the Olympus EM208S
transmission electron microscope. IMMUNOFLUORESCENCE ANALYSIS Immunofluorescence staining with liver tissues or treated cells were performed as we previously reported.12
4′,6-Diamidino-2-phenylindole (DAPI) was used to stain the nucleus in liver tissues and HSCs. All the images were captured with the fluorescence microscope and representative images were
shown. The software Image J was used to quantitate the fluorescent intensity on the micrographs. ANALYSIS OF HSC SENESCENCE HSC senescence was determined by the detection of SA-_β_-gal
(senescence-associated _β_-galactosidase) activity using an SA-_β_-gal staining kit (Cell Signaling). Briefly, adherent cells were fixed with 0.5% glutaraldehyde in PBS for 15 min, washed
with PBS containing 1 mM MgCl2 and stained overnight in PBS containing 1 mM MgCl2, 1 mg/ml X-Gal, 5 mM potassium ferricyanide and 5 mM potassium ferrocyanide. All the images were captured
with a light microscope and representative images were shown. Results were from triplicate experiments. CELL CYCLE ANALYSIS BY FLOW CYTOMETRY Distribution of cell cycle was determined by PI
staining and flow cytometry analysis. HSCs were seeded in six-well plates and cultured in DMEM supplemented with 10% FBS for 24 h, and then were treated with DMSO, Etoposide and DHA at
indicated concentrations for 24 h. Cells were then harvested and fixed, and the cell cycle was then detected by the cellular DNA flow cytometric analysis kit (Nanjing Keygen Biotech)
according to the protocol.22 Percentages of cells within cell cycle compartments (G0/G1, S and G2/M) were determined by flow cytometry (FACS Calibur; Becton, Dickinson and Company, Franklin
Lakes, NJ, USA). The data were analyzed using the software Cell Quest. Results were from triplicate experiments. CALCULATIONS AND STATISTICS Individual culture experiments and animal
experiments were performed in duplicate or triplicate and repeated three times using matched controls, and the data were pooled. Results were expressed as either S.D. or mean±standard error
of the mean (S.E.M.). The statistical significance of differences (*_P_<0.05) was assessed by _t_-test. REFERENCES * Lee YA, Wallace MC, Friedman SL . Pathobiology of liver fibrosis: a
translational success story. _Gut_ 2015; 64: 830–841. Article CAS Google Scholar * Kang N, Gores GJ, Shah VH . Hepatic stellate cells: partners in crime for liver metastases? _Hepatology_
2011; 54: 707–713. Article CAS Google Scholar * Kitano M, Bloomston PM . Hepatic stellate cells and microRNAs in pathogenesis of liver fibrosis. _J Clin Med_ 2016; 5: 10–13. Article
Google Scholar * Lambrecht J, Mannaerts I, van Grunsven LA . The role of miRNAs in stress-responsive hepatic stellate cells during liver fibrosis. _Front Physiol_ 2015; 6: 209. Article
Google Scholar * Thompson AI, Conroy KP, Henderson NC . Hepatic stellate cells: central modulators of hepatic carcinogenesis. _BMC Gastroenterol_ 2015; 15: 63. Article Google Scholar *
Carloni V, Luong TV, Rombouts K . Hepatic stellate cells and extracellular matrix in hepatocellular carcinoma: more complicated than ever. _Liver Int_ 2014; 34: 834–843. Article Google
Scholar * Zhang Z, Guo Y, Zhang S, Zhang Y, Wang Y, Ni W _et al_. Curcumin modulates cannabinoid receptors in liver fibrosis _in vivo_ and inhibits extracellular matrix expression in
hepatic stellate cells by suppressing cannabinoid receptor type-1 _in vitro_. _Eur J Pharmacol_ 2013; 721: 133–140. Article CAS Google Scholar * Zhang F, Zhang Z, Kong D, Zhang X, Chen L,
Zhu X _et al_. Tetramethylpyrazine reduces glucose and insulin-induced activation of hepatic stellate cells by inhibiting insulin receptor-mediated PI3K/AKT and ERK pathways. _Mol Cell
Endocrinol_ 2014; 382: 197–204. Article CAS Google Scholar * Tai X, Cai XB, Zhang Z, Wei R . _In vitro_ and _in vivo_ inhibition of tumor cell viability by combined dihydroartemisinin and
doxorubicin treatment, and the underlying mechanism. _Oncol Lett_ 2016; 12: 3701–3706. Article CAS Google Scholar * Wu C, Liu J, Pan X, Xian W, Li B, Peng W _et al_. Design, synthesis
and evaluation of the antibacterial enhancement activities of amino dihydroartemisinin derivatives. _Molecules_ 2013; 18: 6866–6882. Article CAS Google Scholar * Zhang XG, Li GX, Zhao SS,
Xu FL, Wang YH, Wang W . A review of dihydroartemisinin as another gift from traditional Chinese medicine not only for malaria control but also for schistosomiasis control. _Parasitol Res_
2014; 113: 1769–1773. Article Google Scholar * Zhang Z, Guo M, Zhao S, Shao J, Zheng S . ROS-JNK1/2-dependent activation of autophagy is required for the induction of anti-inflammatory
effect of dihydroartemisinin in liver fibrosis. _Free Radic Biol Med_ 2016; 101: 272–283. Article CAS Google Scholar * Chen Q, Chen L, Kong D, Shao J, Wu L, Zheng S . Dihydroartemisinin
alleviates bile duct ligation-induced liver fibrosis and hepatic stellate cell activation by interfering with the PDGF-betaR/ERK signaling pathway. _Int Immunopharmacol_ 2016; 34: 250–258.
Article CAS Google Scholar * Chen Q, Chen L, Wu X, Zhang F, Jin H, Lu C _et al_. Dihydroartemisinin prevents liver fibrosis in bile duct ligated rats by inducing hepatic stellate cell
apoptosis through modulating the PI3K/Akt pathway. _IUBMB Life_ 2016; 68: 220–231. Article CAS Google Scholar * Xu W, Lu C, Zhang F, Shao J, Zheng S . Dihydroartemisinin restricts hepatic
stellate cell contraction via an FXR-S1PR2-dependent mechanism. _IUBMB Life_ 2016; 68: 376–387. Article CAS Google Scholar * Xu W, Lu C, Yao L, Zhang F, Shao J, Zheng S .
Dihydroartemisinin protects against alcoholic liver injury through alleviating hepatocyte steatosis in a farnesoid X receptor-dependentmanner. _Toxicol Appl Pharmacol_ 2016; 315: 23–34.
Article Google Scholar * Kaul Z, Cesare AJ, Huschtscha LI, Neumann AA, Reddel RR . Five dysfunctional telomeres predict onset of senescence in human cells. _EMBO Rep_ 2012; 13: 52–59.
Article CAS Google Scholar * Shao AW, Sun H, Geng Y, Peng Q, Wang P, Chen J _et al_. Bclaf1 is an important NF-κB signaling transducer and C/EBPbeta regulator in DNA damage-induced
senescence. _Cell Death Differ_ 2016; 23: 865–875. Article CAS Google Scholar * Kagawa S, Natsuizaka M, Whelan KA, Facompre N, Naganuma S, Ohashi S _et al_. Cellular senescence checkpoint
function determines differential Notch1-dependent oncogenic and tumor-suppressor activities. _Oncogene_ 2016; 34: 2347–2359. Article Google Scholar * Vicente R, Mausset-Bonnefont AL,
Jorgensen C, Louis-Plence P, Brondello JM . Cellular senescence impact on immune cell fate and function. _Aging Cell_ 2016; 15: 400–406. Article CAS Google Scholar * Rufini A, Tucci P,
Celardo I, Melino G . Senescence and aging: the critical roles of p53. _Oncogene_ 2013; 32: 5129–5143. Article CAS Google Scholar * Jin H, Lian N, Zhang F, Chen L, Chen Q, Lu C _et al_.
Activation of PPARγ/P53 signaling is required for curcumin to induce hepatic stellate cell senescence. _Cell Death Dis_ 2016; 7: e2189. Article CAS Google Scholar * Romagosa C, Simonetti
S, Lopez-Vicente L, Mazo A, Lleonart ME, Castellvi J _et al_. p16(Ink4a) overexpression in cancer: a tumor suppressor gene associated with senescence and high-grade tumors. _Oncogene_ 2011;
30: 2087–2097. Article CAS Google Scholar * Zhang H, Puleston DJ, Simon AK . Autophagy and immune senescence. _Trends Mol Med_ 2016; 22: 671–686. Article CAS Google Scholar * Kang C,
Elledge SJ . How autophagy both activates and inhibits cellular senescence. _Autophagy_ 2016; 12: 898–899. Article CAS Google Scholar * Lechuga CG, Hernández-Nazara ZH, Hernández E,
Bustamante M, Desierto G, Cotty A _et al_. PI3K is involved in PDGF-beta receptor upregulation post-PDGF-BB treatment in mouse HSC. _Am J Physiol Gastrointest Liver Physiol_ 2006; 291:
G1051–G1061. Article CAS Google Scholar * Bartneck M, Warzecha KT, Tag CG, Sauer-Lehnen S, Heymann F, Trautwein C _et al_. Isolation and time lapse microscopy of highly pure hepatic
stellate cells. _Anal Cell Pathol_ 2015; 2015: 417023. Article Google Scholar * Kim KM, Kim JM, Yoo YH, Kim JI, Park YC . Cilostazol induces cellular senescence and confers resistance to
etoposide-induced apoptosis in articular chondrocytes. _Int J Mol Med_ 2012; 29: 619–624. Article CAS Google Scholar * Truman AW, Kristjansdottir K, Wolfgeher D, Hasin N, Polier S, Zhang
H _et al_. CDK-dependent Hsp70 Phosphorylation controls G1 cyclin abundance and cell-cycle progression. _Cell_ 2012; 151: 1308–1318. Article CAS Google Scholar * Cassidy LD, Narita M .
GATA get a hold on senescence. _Science_ 2015; 349: 1448–1449. Article CAS Google Scholar * El Hasasna H, Athamneh K, Al Samri H, Karuvantevida N, Al Dhaheri Y, Hisaindee S _et al_. Rhus
coriaria induces senescence and autophagic cell death in breast cancer cells through a mechanism involving p38 and ERK1/2 activation. _Sci Rep_ 2015; 5: 13013. Article CAS Google Scholar
* Kim KH, Park B, Rhee DK, Pyo S . Acrylamide induces senescence in macrophages through a process involving ATF3, ROS, p38/JNK, and a telomerase-independent pathway. _Chem Res Toxicol_ 2015;
28: 71–86. Article CAS Google Scholar * Kim JE, Jin DH, Lee SD, Hong SW, Shin JS, Lee SK _et al_. Vitamin C inhibits p53-induced replicative senescence through suppression of ROS
production and p38 MAPK activity. _Int J Mol Med_ 2008; 22: 651–655. CAS PubMed Google Scholar * Fan T, Huang Z, Chen L, Wang W, Zhang B, Xu Y _et al_. Associations between autophagy, the
ubiquitin-proteasome system and endoplasmic reticulum stress in hypoxia-deoxygenation or ischemia-reperfusion. _Eur J Pharmacol_ 2016; 791: 157–167. Article CAS Google Scholar *
Levchenko M, Lorenzi I, Dudek J . The degradation pathway of the mitophagy receptor Atg32 is re-routed by a posttranslational modification. _PLoS One_ 2016; 11: e0168518. Article Google
Scholar * Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L _et al_. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. _Science_ 2015; 349:
aaa5612. Article Google Scholar * Tang BL . mTOR, autophagy, and reprogramming. _Front Cell Dev Biol_ 2014; 1: 4. Article Google Scholar * Maiese K . Targeting molecules to medicine with
mTOR, autophagy and neurodegenerative disorders. _Br J Clin Pharmacol_ 2016; 82: 1245–1266. Article CAS Google Scholar * Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H,
Acevedo Arozena A _et al_. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). _Autophagy_ 2016; 12: 1–222. Article Google Scholar * Krizhanovsky V,
Yon M, Dickins RA, Hearn S, Simon J, Miething C _et al_. Senescence of activated stellate cells limits liver fibrosis. _Cell_ 2008; 134: 657–667. Article CAS Google Scholar * Kong X,
Feng D, Wang H, Hong F, Bertola A, Wang FS _et al_. Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. _Hepatology_ 2012; 56: 1150–1159. Article
CAS Google Scholar * Lentjes MH, Niessen HE, Akiyama Y, de Bruïne AP, Melotte V, van Engeland M . The emerging role of GATA transcription factors in development and disease. _Expert Rev
Mol Med_ 2016; 18: e3. Article Google Scholar * Block DH, Shapira M . GATA transcription factors as tissue-specific master regulators for induced responses. _Worm_ 2015; 4: e1118607.
Article Google Scholar * García-Prat L, Martínez-Vicente M, Perdiguero E, Ortet L, Rodríguez-Ubreva J, Rebollo E _et al_. Autophagy maintains stemness by preventing senescence. _Nature_
2016; 529: 37–42. Article Google Scholar * Liu H, He Z, Simon HU . Autophagy suppresses melanoma tumorigenesis by inducing senescence. _Autophagy_ 2014; 10: 372–373. Article CAS Google
Scholar * Zhang Z, Zhao S, Yao Z, Wang L, Shao J, Chen A _et al_. Autophagy regulates turnover of lipid droplets via ROS-dependent Rab25 activation in hepatic stellate cell. _Redox Biol_
2017; 11: 322–334. Article CAS Google Scholar Download references ACKNOWLEDGEMENTS This work was supported by the National Natural Science Foundation of China (81270514, 31571455,
31401210, 31600653 and 81600483), the Natural Science Foundation of Jiangsu Province (BK20140955), the Open Project Program of Jiangsu Key Laboratory for Pharmacology and Safety Evaluation
of Chinese Materia Medica (JKLPSE 201502), and the Project of the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD). Finally, I sincerely thank my wife
(Mei Guo) for her support and encouragement in a difficult period. I shall love you as long as I have breath. AUTHOR CONTRIBUTIONS SZ and ZZ designed the study. ZZ, ZY, SZ, JS and AC
performed the experiments. ZZ analyzed the data. FZ and SZ contributed to materials and analysis tools. SZ and ZZ prepared the manuscript. SZ provided the financial support. All authors
reviewed and approved the manuscript. AUTHOR INFORMATION Author notes * Zili Zhang, Zhen Yao and Shifeng Zhao: These authors contributed equally to this work. AUTHORS AND AFFILIATIONS *
Department of Pharmacology, School of Pharmacy, Nanjing University of Chinese Medicine, Nanjing, China Zili Zhang, Zhen Yao, Shifeng Zhao, Jiangjuan Shao, Feng Zhang & Shizhong Zheng *
Department of Pathology, School of Medicine, Saint Louis University, St Louis, MO, USA Anping Chen * Jiangsu Key Laboratory for Pharmacology and Safety Evaluation of Chinese Materia Medica,
Nanjing University of Chinese Medicine, Nanjing, China Feng Zhang & Shizhong Zheng * Jiangsu Key Laboratory of Therapeutic Material of Chinese Medicine, Nanjing University of Chinese
Medicine, Nanjing, China Feng Zhang & Shizhong Zheng Authors * Zili Zhang View author publications You can also search for this author inPubMed Google Scholar * Zhen Yao View author
publications You can also search for this author inPubMed Google Scholar * Shifeng Zhao View author publications You can also search for this author inPubMed Google Scholar * Jiangjuan Shao
View author publications You can also search for this author inPubMed Google Scholar * Anping Chen View author publications You can also search for this author inPubMed Google Scholar * Feng
Zhang View author publications You can also search for this author inPubMed Google Scholar * Shizhong Zheng View author publications You can also search for this author inPubMed Google
Scholar CORRESPONDING AUTHOR Correspondence to Shizhong Zheng. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no conflict of interest. ADDITIONAL INFORMATION Edited by B
Zhivotovsky Supplementary Information accompanies this paper on Cell Death and Disease website SUPPLEMENTARY INFORMATION SUPPLEMENTARY FIGURE LEGENDS (DOCX 25 KB) SUPPLEMENTARY FIGURE S1
(JPG 182 KB) SUPPLEMENTARY FIGURE S2 (JPG 154 KB) SUPPLEMENTARY FIGURE S3 (JPG 2043 KB) SUPPLEMENTARY FIGURE S4 (JPG 1156 KB) SUPPLEMENTARY FIGURE S5 (JPG 284 KB) SUPPLEMENTARY FIGURE S6
(JPG 1008 KB) RIGHTS AND PERMISSIONS _Cell Death and Disease_ is an open-access journal published by _Nature Publishing Group_. This work is licensed under a Creative Commons Attribution 4.0
International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the
material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/ Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Zhang, Z., Yao, Z., Zhao, S. _et al._ Interaction between autophagy and senescence
is required for dihydroartemisinin to alleviate liver fibrosis. _Cell Death Dis_ 8, e2886 (2017). https://doi.org/10.1038/cddis.2017.255 Download citation * Received: 17 April 2017 *
Revised: 17 April 2017 * Accepted: 03 May 2017 * Published: 15 June 2017 * Issue Date: June 2017 * DOI: https://doi.org/10.1038/cddis.2017.255 SHARE THIS ARTICLE Anyone you share the
following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer
Nature SharedIt content-sharing initiative