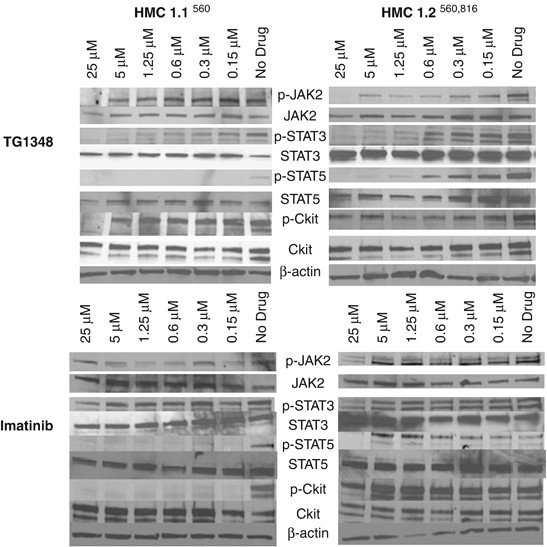
Inhibition of JAK–STAT signaling by TG101348: a novel mechanism for inhibition of KITD816V-dependent growth in mast cell leukemia cells
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

Systemic mastocytosis (SM) is a stem-cell-derived clonal myeloproliferation characterized by an accumulation of abnormal mast cells. A majority of SM patients harbor the KITD816V mutation,
indicating its potentially important role in disease pathogenesis. KITD816V activates several downstream signaling pathways including PI3-kinase/AKT, STAT5 and ERK-1/2 that mediate its
proliferative, survival and differentiation effects. SRC family kinases have an important role in stem cell factor (SCF)-induced cell proliferation; in SCF-responsive cell lines and
hematopoietic progenitor cells, LYN is associated with the juxtamembrane region of KIT, and is rapidly phosphorylated in response to SCF stimulation.1 Coexpression of KIT with functionally
defective CBL, normally a negative regulator of KIT, in murine bone marrow cells leads to generalized mastocytosis.2 Interestingly, KIT kinase activity was dispensable for cell
transformation mediated by mutant CBL; instead, transformation was dependent on the SRC family kinase, FYN. Furthermore, recent data suggest that SRC kinase activity is important for full
expression of KITD816V's transforming potential; KITD816V not only activates receptor kinase activity, but also subverts its substrate specificity to confer SRC-like kinase activity.3 JAK2
also appears to be an integral component of the SCF/KIT signaling pathway; in SCF-responsive MO7e cells, JAK2 is constitutively associated with KIT in unstimulated cells; SCF stimulation
results in recruitment of JAK2 to the KIT receptor complex, and induces rapid phosphorylation of the former.4 Furthermore, JAK2 knockdown with antisense oligonucleotides resulted in marked
inhibition of SCF-induced cell proliferation, thereby confirming its key role in the SCF/KIT signaling cascade.
Current therapy of SM is suboptimal and includes interferon-α and 2-chlorodeoxyadenosine, which are capable of achieving mast cell cytoreduction (reviewed by Pardanani and Tefferi5).
Although several drugs have demonstrable anti-KIT activity in vitro, their clinical benefit to date has been limited. Imatinib mesylate, a KIT inhibitor, does not inhibit KITD816V, but has
activity against other rare SM-relevant KIT mutations, such as KITF522C (transmembrane domain) and KITV560G (juxtamembrane domain); consequently, it appears to have a limited role for the
treatment of adult SM. Dasatinib inhibits both juxtamembrane and activation-loop KIT mutants in vitro, and in addition has anti-SRC activity; it has limited activity, however, in the
treatment of SM patients and clinical responses are mostly limited to alleviation of symptoms.
AP is partly supported by a grant from the Henry J Predolin Foundation. TL generated and analyzed the laboratory data; AP and AT designed the study, analyzed the laboratory data and wrote
the paper.
Division of Hematology, Department of Medicine, Mayo Clinic, Rochester, MN, USA
Anyone you share the following link with will be able to read this content: