Tiam1/rac1 complex controls il17a transcription and autoimmunity
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT RORγt is a master transcription factor of Th17 cells and considered as a promising drug target for the treatment of autoimmune diseases. Here, we show the guanine nucleotide
exchange factor, Tiam1, and its cognate Rho-family G protein, Rac1, regulate interleukin (IL)17A transcription and autoimmunity. Whereas _Tiam1_ genetic deficiency weakens IL-17A expression
partially and inhibits the development of experimental autoimmune encephalomyelitis (EAE), deletion of _Rac1_ in T cells exhibits more robust effects on Th17 cells and EAE. We demonstrate
Tiam1 and Rac1 form a complex with RORγt in the nuclear compartment of Th17 cells, and together bind and activate the _Il17_ promoter. The clinical relevance of these findings is emphasized
by pharmacological targeting of Rac1 that suppresses both murine and human Th17 cells as well as EAE. Thus, our findings highlight a regulatory pathway of Tiam1/Rac1 in Th17 cells and
suggest that it may be a therapeutic target in multiple sclerosis. SIMILAR CONTENT BEING VIEWED BY OTHERS GENETIC AND PHARMACOLOGICAL INHIBITION OF THE NUCLEAR RECEPTOR RORΑ REGULATES TH17
DRIVEN INFLAMMATORY DISORDERS Article Open access 04 January 2021 SOX-5 ACTIVATES A NOVEL RORΓT ENHANCER TO FACILITATE EXPERIMENTAL AUTOIMMUNE ENCEPHALOMYELITIS BY PROMOTING TH17 CELL
DIFFERENTIATION Article Open access 20 January 2021 PPARΑ KNOCKOUT IN MICE INCREASES THE TH17 DEVELOPMENT BY FACILITATING THE IKKΑ/RORΓT AND IKKΑ/FOXP3 COMPLEXES Article Open access 14 July
2023 INTRODUCTION T helper (Th)17 cells are considered to play a pivotal role in the pathogenesis of multiple sclerosis (MS) as well as its animal model, experimental autoimmune
encephalomyelitis (EAE)1,2. Naive CD4+ T cells differentiate into Th17 cells when activated in the presence of transforming growth factor (TGF)-β and interleukin (IL)-6 (ref. 3). Alongside
their signature cytokines, IL-17A and IL-17F, Th17 cells are characterized by their expression of pro-inflammatory cytokines such as IL-22 and granulocyte–macrophage colony-stimulating
factor (GM-CSF)4,5. The pro-inflammatory function of IL-17A is demonstrated by the fact that IL-17A deficient mice were protected from EAE6. IL-17A neutralization is a promising therapy for
Th17-associated autoimmune diseases such as psoriasis, ankylosing spondylitis and MS7,8,9. Recent success in clinical trials for the treatment of psoriasis and rheumatoid arthritis with
biologics that inhibit the IL17A-IL17R axis (Ixekizumab and Brodalumab) further underscores the importance of this pathway in human autoimmunity10,11,12. The transcription factor RAR-related
orphan receptor gamma (RORγt), recognized as the master transcription factor of Th17 cells, promotes Th17 cell differentiation and is essential for the development of murine and human Th17
cells13,14. RORγt deficient mice are resistant to autoimmune diseases13. RORγt functions in concert with IL-6/STAT3, TGFβ1, and IL-23 to drive the generation of pathogenic Th17
cells15,16,17. RORγt also belongs to the nuclear hormone receptors (NHRs), a well characterized family of transcription factors composed of modular protein structures comprising DNA- and
ligand-binding domains (DBDs and LBDs). While DBDs confer gene target site specificity, LBDs act as control switches for NHR function18. The RORγt LBD is therefore an ideal domain that can
be targeted via small molecules. Numerous studies have identified the downstream genomic targets of RORγt in CD4+ T cells19,20,21, however, very little is known about endogenous ligands that
control RORγt function in Th17 cells. Rho-GTPases such as Rac1 function as molecular switches that cycle between active GTP-bound and inactive GDP-bound states. In their active state, they
interact with effector molecules and stimulate signalling pathways controlling cytoskeletal dynamics, membrane trafficking and gene expression programs22,23. As a well characterized
membrane-bound signal transducing molecule, Rac1 is involved in regulating cell motility and adhesion in addition to the progression of the cell cycle, mitosis, cell death and gene
expression24. Since an elevated level of expression and activity of this protein has been associated with cancer metastasis, direct regulation of Rac1 activity is a potential strategy
employed in the treatment of certain cancers25. Rac1 regulates several signalling pathways in cancer cells including the Wnt/β-catenin pathway by stimulating the assembly of
β-catenin-lymphoid enhancer factor-1 complex26. T lymphoma invasion and metastasis 1 (Tiam1) is a guanine nucleotide exchange factor (GEF) of Rac1 that is believed to act as an oncogene27.
Acting principally upstream of Rac1, Tiam1 is mainly involved in the regulation of Rac1-mediated signalling pathways including cytoskeletal activities, endocytosis and membrane trafficking
as well as cell polarity, migration, adhesion, carcinogenesis and metastasis28,29. Together, the Tiam1/Rac1 complex constitutes a critical component in the biology of human tumours, in both
transformed cells and the accessory cells of the tumour microenvironment30,31. In the present study, we investigate the role of Tiam1/Rac1 signalling in mediating murine and human Th17 cell
development and altering cytokine expression profile. Using genetic mouse models as well as small molecule inhibitors, we identify a novel role of the Tiam1/Rac1 complex in the regulation of
RORγt-mediated _Il17a_ transcription and autoimmune inflammation. RESULTS INCREASED EXPRESSION OF TIAM1 AND RAC1 IN TH17 CELLS We investigated a possible role of the Tiam1/Rac1 complex in
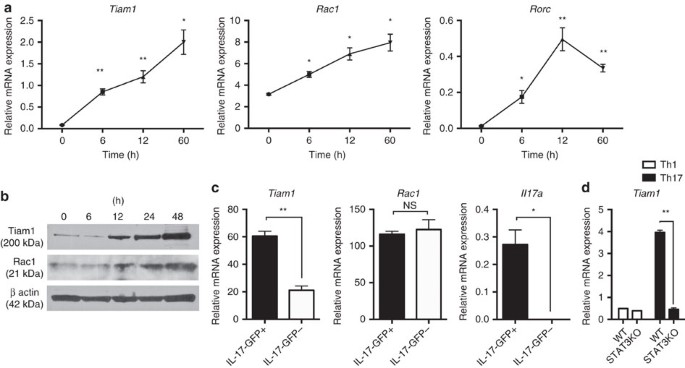
Th17 cells. We found that in Th17 cells, Tiam1 expression is induced within 6 h after polarizing naive CD4+CD62LhiCD44low cells with TGF-β1 and IL-6 as measured at the gene and protein
levels (Fig. 1a,b; Supplementary Fig. 1). Moreover, Rac1 expression was detected in naive CD4+ T cells and was further increased at both the mRNA and protein levels (Fig. 1a,b). To ascertain
the specificity of high Tiam1 expression among Th17 cells, we investigated the relative expression of Tiam1 in CD4+ T cells isolated from myelin oligodendrocyte glycoprotein peptide
(MOG)35–55-immunized IL-17A-GFP knock-in mice 10 days after immunization. IL-17A-GFP+ and IL-17A-GFP− cells were separated by fluorescence-activated cell sorting (FACS) and Tiam1 expression
was analyzed by quantitative PCR. Tiam1 was increased nearly by threefolds in IL-17A-GFP+ lymphocytes compared with the IL-17A-GFP− counterpart, while Rac1 was constitutively expressed at
the gene level in both cell populations (Fig. 1c). Thus, the guanine nucleotide exchange factor, Tiam1, is specifically expressed in IL-17A-producing CD4+ T cells. Given the rapid induction
of Tiam1 in Th17 cells, we sought to investigate molecular signalling pathways that are involved in Tiam1 transcription. STAT3, a key downstream mediator of IL-6 signalling, binds the IL-17
promoter and induces RORγt production32. To study the potential role of STAT3 in inducing Tiam1 expression, we extracted high-resolution transcription factor-DNA interaction profiles from
chromatin immunoprecipitation and high-throughput sequencing (ChIP-seq) data deposited on Gene Expression Omnibus (GEO)32. STAT3 demonstrated high-confidence binding to the promoter region
of all the Tiam1 isoforms (Supplementary Fig. 2). To study whether this interaction is functional, we took advantage of the STAT3 conditional knockout mouse model. Naïve CD4+ T cells from
_CD4-cre x STAT3_fl/fl mice and _STAT3_fl/fl littermates were differentiated under Th17 polarizing conditions and Tiam1 expression was assessed 24 h after _in vitro_ stimulation. We found
that STAT3 deficient Th17 cells expressed significantly lower Tiam1 levels than their wild-type (WT) counterpart (Fig. 1d). TIAM1 DEFICIENCY PARTIALLY REDUCES IL-17A EXPRESSION AND EAE Given
the induction of Tiam1/Rac1 in Th17 cells, we investigated the role of the GEF, Tiam1, in IL-17 expression. Naive CD4+ T cells purified from splenocytes of _Tiam1−_/− mice and their control
WT littermates (_Tiam1+_/+) were differentiated for 4 days under Th1, Th2, Th17 and iTreg conditions and then analyzed by intracellular cytokine staining. We found that genetic deletion of
Tiam1 moderately reduced IL-17A production and increased IL-9 and IL-10, while IFNγ expression was not altered in Th17 cells (Fig. 2a). This phenomenon was specific to Th17 cells since
cytokine profile of CD4+ T cells polarized _in vitro_ under Th1, Th2 and iTreg conditions were not influenced by Tiam1 genetic deletion (Fig. 2b). The modest change in cytokine expression in
Th17 cells was also reflected at the gene level with a twofold reduction in _Il17a_ production and a notable increase in _Il9_ and _Il10_ expression in _Tiam1−_/− cells (Fig. 2c). Since
Tiam1 appears to contribute moderately to the IL-17A-producing capacity of Th17 cells, we were interested in investigating how this translates _in vivo_ at the disease level. EAE was induced
in _Tiam1−_/− and WT _Tiam1+_/+ control mice by immunization with MOG35–55 in complete Freund adjuvant (CFA) that was administered subcutaneously and followed by intraperitoneal (i.p.)
injection of pertussis toxin (PT). We found that EAE induction was delayed in the _Tiam1−_/− mice compared with the _Tiam1+_/+ littermates and the disease course was milder (Fig. 2d). This
delay is statistically significant particularly when analyzed from disease onset until 15 days post-immunization (_P_<0.0015 by two-tailed Mann–Whitney _U_ test) (Fig. 2d). Analysis of
the cytokine profile of MOG35–55-reactive CD4+ T cells was carried out by Enzyme-Linked ImmunoSpot (ELISPOT) technique as well as by Luminex bead-based assay. Accordingly, spleen cells were
isolated from immunized mice 8–10 days after immunization and cells were exposed to MOG35–55 peptides (10 μg ml−1) for 36 h. We found that splenocytes from _Tiam1−_/− mice produced
significantly less IL-17A and IFNγ compared with _Tiam1+_/+ control mice (Fig. 2e). This was confirmed by Luminex assay where the release of IL-17A and IFNγ in the culture supernatants was
lower in MOG35–55-reactive T cells from _Tiam1−_/− mice. We also observed a decrease in IL-22 and GM-CSF, while IL-9 and IL-10 were slightly but not significantly upregulated in the absence
of Tiam1 (Fig. 2f). Given that _Tiam1−_/− mice are a germline knockout mouse model that doesn't distinguish between the effects of Tiam1 deficiency in T cells from its deficiency in
antigen-presenting cells (APCs), it is possible that the observed decrease in IFNγ production could be linked to both T-cell-dependent and independent mechanisms. To measure the influence of
Tiam1 on the magnitude of myelin-specific T-cell response, we analyzed MOG35–55-induced spleen cell proliferation in a criss-cross experiment. Thus, we isolated CD4+ T cells and CD11c+
dendritic cells from spleens of _Tiam1−_/− and _Tiam1+_/+ mice 10 days after immunization and cells were activated with MOG35–55 peptide for two days followed by quantitation of cell
proliferation using [3H]-thymidine incorporation method. We found that although Tiam1 deficiency in CD11c+ cells reduced slightly but not significantly their antigen-presenting function,
lack of Tiam1 in CD4+ T cells had marked effects on their proliferation (Fig. 2g). To establish a more direct comparison of the encephalitogenicity of _Tiam1−_/− versus WT donor T cells, we
utilized an adoptive transfer EAE model. Draining lymph nodes (LNs) from MOG35–55/CFA-immunized _Tiam1−_/− and _Tiam1+_/+ were re-activated _in vitro_ with MOG35–55 peptide plus recombinant
IL-23 in the presence of WT dendritic cells according to established protocols for 2 days33 followed by transfer of equal numbers of CD4+ T cells replace in with to Rag1_−_/− lymphopenic
mice that were monitored for clinical disease development. We found that EAE severity was slightly but significantly reduced in recipients of Tiam1-deficient T cells compared with WT T cells
(mean maximal score for _Tiam1−_/− T cells compared with WT T cells between days 11 and 15 post-transfer; _P_<0.05 by Mann–Whitney _U_ test; _n_=5/group) (Fig. 2h) leaving room for
exploration of the downstream GTPase, Rac1. RAC1 IS REQUIRED FOR IL-17A EXPRESSION AND INDUCTION OF EAE GEF’s such as Tiam1 are required for activation of the GTPase Rac1 that has been
implicated in a myriad of cellular processes. We investigated the role of Rac1 in the generation of Th17 cells _in vitro_ and _in vivo_. Floxed Rac1 (_Rac1_fl/fl) mice were crossed with
CD4-Cre syngeneic mice to generate animals with conditional deletion of Rac1 in CD4+ T cells (_CD4-Cre x Rac1_fl/fl). Naive CD4+ T cells from _CD4-Cre x Rac1_fl/fl as well as _Rac1_fl/fl
control mice were differentiated for 4 days under Th1, Th2 and Th17 cell polarization conditions. Interestingly, CD4+ cells from _CD4-Cre x Rac1_fl/fl mice had a specific and significant
decrease in IL-17A production accompanied by an increase in IL-9 and IL-10 frequency under Th17 conditions induced by IL-6 plus TGF-β1, with no significant change in cytokines under Th1 and
Th2 polarizing conditions (Fig. 3a). Further analysis of cytokine expression of differentiated Th17 cells by Taqman PCR confirmed a marked decrease in _Il17a_ expression that was associated
with an increase in _Il9_ and to a lesser extent _Il10_ expression. Rac1 deficiency in T cells does not modulate _Rora_/_Rorc_ or other Th17-associated molecules (Fig. 3b). To test the
effects of Rac1 on the differentiation of Th17 cells in the presence of IL-23, naive CD4+ T cells from Rac1-deficient mice and control littermates were differentiated for 4 days under Th17
conditions of IL-6 plus TGF-β1, rested for 2 days, and then cultured in the presence of recombinant IL-23 for 2 more days. Cytokine analysis of the culture supernatants showed that Rac1
deletion reduced significantly IL-17A in addition to a reduction in IL-21, IL-22 and GM-CSF (Fig. 3c). In addition, we showed that Th17 cells with Rac1 deficiency differentiated using IL-1β,
IL-6, and IL-23 exhibited a decrease in IL-17A, IL-17F, IL-22 and IL-23 receptor expression (Supplementary Fig. 3). Next, we examined the function of Rac1 in EAE mice. We found that
clinical scores in _CD4-Cre x Rac1_fl/fl mice were significantly reduced compared with _Rac1_fl/fl control littermates. The mean maximal score of _CD4-Cre x Rac1_fl/fl mice was 2.5±0.3
versus 4.1±0.4 in control _Rac1_fl/fl, _P_=0.008 by Mann–Whitney _U_ test (Fig. 3d). Analysis of cytokine production of MOG35–55-reactive CD4+ T cells in spleen and LN tissues by ELISPOT
analysis showed a significant decrease in IL-17A-reactive cells in _CD4-Cre x Rac1_fl/fl mice compared with control mice while IFNγ production was not significantly changed (Fig. 3e for
spleen and Fig. 3f for LN). A panel of cytokines was measured by Luminex in spleen and LN cells treated with MOG35–55 peptide, which further demonstrates the involvement of Rac1 in IL-17A
production in addition to other Th17-associated cytokines such as IL-21, IL-22 and GM-CSF (Supplementary Fig. 4). Although not significant, a slight increase in IL-9 expression was observed
in Rac1 deficient mice, suggesting that IL-9 production is not a major player in the disease course of _CD4-Cre x Rac1_fl/fl mice (Supplementary Fig. 4). Finally, we found that Rac1 deletion
reduced the ability of MOG35–55 stimulation to induce T-cell proliferation (Fig. 3g). To analyze the cytokine profile of CD4+ T cells in the central nervous system (CNS) of EAE mice, spinal
cord tissues were collected 12 days after immunization, and infiltrated cells were isolated and processed for cytokine expression by Taqman PCR. Our findings confirm that Rac1 deletion
causes a decrease in the expression of _Il17a_, _Il21_ and _Il22_. Interestingly, a decrease in _Csf2_ (gene encoding for GM-CSF) and to a lesser extent _Ifng_ was detected in the spines of
_CD4-Cre x Rac1_fl/fl mice (Fig. 3h). In addition, histological examination revealed marked decrease in inflammatory infiltrates in spinal cord sections of _CD4-Cre x Rac1_fl/fl mice
compared with control _Rac1_fl/fl mice (Fig. 3i). TIAM1/RAC1 INHIBITION REDUCES IL-17A AND EAE SYMPTOMS Our findings suggest that targeting the _Tiam1_/_Rac1_ complex is a viable approach
for regulating Th17 cell-mediated autoimmune encephalomyelitis _in vivo_. We tested Tiam1/Rac1 inhibition in Th17 cells polarized with IL-6 plus TGF-β1 _in vitro_ using NSC23766, a small
molecule that effectively inhibits the Rac1-specific GEF, Tiam1, from binding and activating Rac1 (ref. 34). Our results showed that NSC23766-treated Th17 cells had a marked reduction of
IL-17 producing T cells (Fig. 4a). Analysis of Th17 cells by Taqman showed a decrease in the mRNA expression of IL-17A, IL-17F and IL-23R, whereas IL-9 expression was elevated. Treatment
with NSC23766 did not affect IFNγ, Tbx21 and GM-CSF expression that were weakly expressed in Th17 cells differentiated with IL-6 plus TGF-β1 (Fig. 4b). There was no effect of NSC23766 on
cytokine expression by Th1, or Th2 cells (Fig. 4a). We did not observe any modulation of cell activation or survival of CD4+ T cells treated _in vitro_ with NSC23766 (Supplementary Fig.
5A,B). _In vivo_, administration of eight consecutive injections of NSC23766 (5 mg kg−1, i.p. daily) daily i.p. during the priming phase of EAE (days 0–7 post-immunization) alleviated
clinical disease (mean maximal score of NSC23766-treated mice 1.9±0.4 versus 3.7±0.3 in control phosphate-buffered saline (PBS) recipients, _P_=0.01 by two-tailed Mann–Whitney _U_ test)
(Fig. 4c). The protective effects of NSC23766 were accompanied by a significant decrease in MOG35–55-reactive IL-17A-producing CD4+ T cells measured in the splenocytes by ELISPOT with no
significance alterations of IFNγ-producing T cells (Fig. 4d). To measure the influence of NSC23766 on the magnitude of MOG35–55-specific T-cell response, we measured splenocyte proliferation
of control and treated mice. Splenocytes were re-challenged _in vitro_ with increasing doses of MOG35–55 peptide (0–20 μg ml−1) and cell proliferation was measured 2 days later. We found
that NSC2366-treated mice exhibit decreased cell proliferation (Fig. 4e). Given that Tiam1/Rac1 signalling is implicated in cell migration, we observed a notable reduction in CD4+ T-cell
infiltration in the spinal cord tissues in mice that received NSC23766 treatment. This suggests that in addition to the reduction in IL-17+ CD4+ T cells by NSC23766 treatment, decreased
T-cell infiltration may contribute to the protective effects of NSC23766 in EAE (Supplementary Fig. 6). To confirm the protective effects of Rac1 pharmacological inhibition in EAE, we
utilized EHT1864 another small-molecular weight compound that directly inhibits active Rac1 (ref. 35). Mice received eight consecutive injections of EHT1864 (40 mg kg−1) every day according
to the regimen described above and were monitored for clinical symptoms for up to 30 days after immunization. As expected, inhibition of Rac1 activity ameliorated disease severity confirming
the therapeutic effects of targeting Rac1 in EAE (mean maximal score of EHT1864-treated mice 1.7±0.4 versus 3.4±0.3 in control PBS recipients, _P_=0.009 by two-tailed Mann–Whitney _U_ test)
(Fig. 4f). In agreement with the data reported in the Rac1-deficient mice, we found that pharmacological inhibition of Rac1 activation decreased significantly MOG35–55-reactive
IL-17A-producing CD4+ T cells as measured in the splenocytes by ELISPOT with no significant alterations of IFNγ-producing T cells (Fig. 4g). To measure the influence of EHT1864 on the
magnitude of MOG35–55-specific T-cell response, we measured splenocyte proliferation of control and treated mice. We found that splenocytes from EHT1864-treated mice exhibited decreased cell
proliferation (Fig. 4h). TIAM1/RAC1 AND RORΓT COOPERATIVELY BIND TO IL17 PROMOTER Differentiation of Th17 cells is controlled by the master transcription factor, RORγt, which dictates a
specific and heritable gene expression profile13. Given that Rac1 has been shown to modulate gene expression in various cell types23, we hypothesized that the Tiam1/Rac1 complex may regulate
Th17 cell differentiation by direct interaction with RORγt. We performed temporal and spatial protein–protein binding studies in Th17 cells on per cell basis using proximity ligation assay
(PLA), a very sensitive technique to measure protein–protein interaction as detailed in ‘Methods’ section. First, we found that Tiam1 and Rac1 interact physically in Th17 cells starting at 6
h and this interaction is further enhanced at 24 and 48 h after differentiation, in both the cytoplasmic and nuclear compartments (Fig. 5a–c). Interestingly, we found that Tiam1 interacting
with RORγt in Th17 cells was detected mostly in the cytoplasm 6 h after differentiation followed by accumulation of the Tiam1/RORγt complex in the both the nuclei and cytoplasm of Th17
cells at 24 and 48 h (Fig. 5d–f) In a second set of experiments, co-immunoprecipitation (co-IP) studies were performed using cytoplasmic and nuclear extracts from Th17 cells polarized for 48
h to confirm the observed interaction between endogenous Tiam1/Rac1 and RORγt. Precipitation with anti-Tiam1 antibody yielded bands corresponding to both Rac1 and RORγt, whereas
precipitation with the nonspecific IgG antibody serving as the negative control yielded neither band (Fig. 5g; Supplementary Fig. 7). Specifically, we observed a strong interaction between
Tiam1 and Rac1 in both extracts whereas Tiam1/RORγt interaction was mainly detected in the nuclear extracts. Taken together, our findings demonstrate the existence of a Tiam1_–_Rac1–RORγt
complex in Th17 cells. Next, we assessed whether the Tiam1/Rac1 complex is recruited to the _Il17_ promoter in Th17 cells. Naive CD4+ T cells were differentiated for 48 h and chromatin
immunoprecipitation followed by PCR (ChIP-PCR) analysis was used to measure a possible recruitment of Tiam1 and/or Rac1 to the conserved non-coding sequence (CNS2) of _Il17_ promoter, a
_cis_ element that physically interacts with both _Il17a_ and _Il17f_ gene promoters and is required for RORγt-driven IL-17 production in Th17 cells36. ChIP assay using PCR primers
encompassing the _CNS2_ region demonstrated the suspected dual involvement of Tiam1 and Rac1 in RORγt-mediated binding specifically to the CNS2 of the _Il17_ promoter (Fig. 5h). First, we
found that both Tiam1 and Rac1 are present at the CNS2 48 h after Th17 cell differentiation. Note the high degree of Tiam1/Rac1 nuclear translocation at the same time point (Fig. 5a,g).
Second, pharmacological inhibition of Rac1 using NSC23766 not only abolished Tiam1 and Rac1 binding to the CNS2 but also it has hindered RORγt recruitment to the _Il17_ promoter (Fig. 5h).
Third, using conditional RORc null mice (_CD4-Cre x RORc_fl/fl), we found that the recruitment of Tiam1/Rac1 complex to CNS2 is completely abolished in the absence of RORγt in Th17 cells
(Fig. 5i). Altogether, these findings suggest that Tiam1/Rac1 nuclear translocation and interaction with the CNS2 region in Th17 cells is RORγt-dependent. To analyze the functional relevance
of Tiam1/Rac1 physical interaction with RORγt in Th17 cells, we investigated the ability of Tiam1-mediated Rac1 activation to induce the _Il17_ promoter in reporter assays using 293T cells.
We used the reporter construct pGL3-CNS2, containing the firefly luciferase gene under the control of the _Il17_ promoter described previously36. Co-transfection of the pGL3-CNS2 luciferase
reporter construct with a plasmid encoding the N-terminal truncated mutant of Tiam1 (C1199-Tiam1), a Tiam1 variant that has been shown to be more stable and more active than the full
length37, resulted in a significant amplification of RORγt-induced _Il17_ promoter activation (_P_<0.01 by unpaired Student’s _t_-test) (Fig. 5j). This was significantly but not totally
inhibited when transfected cells were treated with the Rac1 inhibitor NSC23766 (_P_<0.01 by unpaired Student’s _t_-test) (Fig. 5j). The fact that NSC23766 only partially inhibits
Tiam1-mediated _Il17_ promoter activation suggests the other GEFs may be involved in the induction of Rac1 activation in this system. The specific involvement of Tiam1 in the enhancement of
RORγt-induced _Il17_ promoter activation is further verified by the lack of Tiam1-mediated regulation of the function of T-bet, the master transcription factor of Th1 cells. Indeed, we found
that Tiam1 does not regulate _Ifng_ promoter activation when overexpressed alone or in the presence of T-bet in a luciferase reporter assay (Supplementary Fig. 8). This is in agreement with
the findings that inhibition of Tiam1 and Rac1 in Th1 cells exhibited only modest effects on IFNγ production (Figs 2a and 3a). HUMAN TIAM1/RAC1 PHARMACOLOGICAL INHIBITION DECREASES IL-17A
Our findings that Rac1 inhibition suppressed murine Th17 cell differentiation and EAE development led us to question whether Rac1 signalling is also involved in human T-cell development.
Therefore, we analyzed Tiam1 and Rac1 expression in human Th17 cells. Compared with Th1 cells, we found that the expression of both Tiam1 and Rac1 is significantly higher in Th17 cells
differentiated _in vitro_ according to a standard protocol for 7 days. In addition, pharmacological inhibition of Tiam1/Rac1 activity using NSC23766 reduced IL-17A mRNA expression in Th17
cells without regulating _Rorc_ gene level suggesting that NSC23766 effects are independent of RORγt expression (Fig. 6a). To further evaluate the role of Tiam1/Rac1 in T cells, CD4+ T cells
were differentiated under Th1 and Th17 cell conditions in the presence or absence of NSC23766. Analysis of IL-17A expression by flow cytometry demonstrated that NSC23766 specifically
regulates IL-17A expression in Th17 cells while sparing IFNγ in both Th1 and Th17 cells (Fig. 6b,c; Supplementary Fig. 9A). In agreement with the data generated in mouse Th17 cells, NSC23766
downregulated the production of IL-22 and GM-CSF in addition to inducing a robust decrease in IL-17A. In contrast, addition of NSC23766 upregulated IL-9 and IL-10 as measured by bead-based
Luminex assay in Th17 cell cultures (Fig. 6d). Notably, we detected a significant decrease in soluble IL-2Rα, a pro-inflammatory cytokine that has been shown to enhance the development of
Th17 cells and to exacerbate autoimmunity in EAE mice38 (Fig. 6d). To determine whether Tiam1 and Rac1 expression is altered in the inflammatory conditions of MS patients, we extracted gene
microarray data of CD4+ T cells from eight MS patients and four healthy controls deposited on GEO (accession code GSE32988)39. Strikingly, there was a significant increase in Tiam1 and Rac1
gene expression in CD4+ T cells from MS patients compared with healthy controls (Fig. 6e). Of interest, we noticed a dominant Th17 cytokine profile in this group of MS patients as revealed
by increased expression of _Il17a_, _Il17f, Il21_, _IL23r_ and _Rorc_ (Fig. 6e; Supplementary Fig. 9B). These findings further highlight a possible involvement of Tiam1/Rac1 signalling in
Th17 cell development in MS patients. DISCUSSION The master transcription factor of Th17 cells, RORγt, is a well characterized drug target that holds promise for autoimmune disease therapy.
Although several groups have reported on small molecule inhibitors of RORγt that are capable of binding to its LBD and suppressing _Il17_ transcription _in vitro_ and _in vivo_18,21, the
regulation of this orphan nuclear receptor by endogenous proteins has yet to be determined. Our present study identifies a novel signalling pathway where the Tiam1/Rac1 complex regulates
RORγt function in Th17 cells. Most of the cellular functions attributed to the Tiam1/Rac1 complex depend on its spatio-temporal-controlled activation. Whereas it is generally accepted that
Tiam1/Rac1 signals at the plasma membrane or on intracellular vesicles, there is increasing evidence for GTPase signalling in the nucleus40,41. This is further supported by the discovery
that some GTPases such as Rac1 possess a nuclear localization signal raising the possibility that Rac1 may play an important role not only in the cytosol but in the nucleus as well31. Our
confocal and ChIP analyses provide novel evidence that both Tiam1 and Rac1 are induced in the cytoplasm and translocate to the nucleus in Th17 cells where they are recruited to the _Il17_
promoter in a RORγt-dependent manner as evidenced by the use of RORc-deficient Th17 cells. Protein translocation across the nuclear membrane occurs through nuclear pore complexes that assume
the role of gatekeepers. Small molecules (<40 kD) such as Rac1 are able to pass through the nuclear pore by diffusion, while active nuclear import of protein complexes such as Tiam1/Rac1
is mediated by karyopherins42. Whether RORγt is playing a direct role in the nuclear translocation of Rac1/Tiam1 complex remains to be determined. Given the large number of protein
interaction domains found in its structure, Tiam1 has been described to associate with a myriad of membrane and intracellular proteins such as CD44 (ref. 43), spinophilin44, and JIP/IB2
complex45 thus regulating downstream gene transcription. These reported findings are in agreement with our present study of Th17 cells where Tiam1 co-localizes with RORγt in the nucleus of
Th17 cells as we demonstrated by confocal images and further confirmed by ChIP data at the _Il17_ promoter level. Pathogenic subpopulations of Th17 cells are characterized by co-expression
of RORγt and T-bet, co-produce IL-17 and IFN-γ, and drive autoimmunity in mice46. They are distinct from ‘classical’ Th17 cells that are not pathogenic, and produce mainly IL-17, IL-9 and
IL-10 (ref. 15). We find that inhibition of Tiam1/Rac1 activation regulates the cytokine profile of Th17 cells as evidenced by a down-regulation of IL-17A and an up-regulation in IL-9 and
IL-10 expression at the gene and protein levels. Previously, we have reported that Th17 cells lacking RORγt expression exhibit a cytokine signature similar to that found in non-pathogenic
Th17 cells, namely a decrease in IL-17A and IL-17F and an increase in IL-9 and IL-10 expression20. Our present _in vivo_ findings showing amelioration of EAE clinical disease in response to
genetic deletion or pharmacological inhibition of Tiam1/Rac1 activity suggest a potential role of Tiam1/Rac1 not only in the generation of IL-17A-producing CD4+ T cells, (IL-17 being a key
cytokine required for EAE induction47), but also in potentiating their encephalitogenicity. Given that inhibition of the Tiam1/Rac1 axis confers a similar phenotype to that reported in
RORγt-deficient Th17 cells, further supports that RORγt downstream signalling is a critical target of the Tiam1/Rac1 complex. Having been mostly investigated in the context of neoplasms,
targeting the Tiam1/Rac1 complex either genetically or using pharmacological inhibitors has shown the ability to deter proliferative ability of several tumours30,48 in addition to primary
cells49,50. The effects of Rac1 on Th17 cell development are postulated to go beyond the transcriptional effects on IL-17A to encompass the ability of these cells to migrate and form
efficient immune synapses with APCs. The pronounced anti-proliferative effects mediated by genetic deficiency or pharmacological inhibition of Tiam1/Rac1 complex might be rooted in the
hindered ability of T cells to establish optimal contact with APCs, which is needed to perpetuate T-cell receptor activation and cellular proliferation. Rho-GTPases including Rac1 have been
shown to play a crucial role in cytoskeletal remodelling that mediates several cellular processes such as cellular migration, polarization and efficient immunological synapse formation51. As
such, active Rac1 orchestrates actin polymerization needed for the formation of lamellopodia, fillopodia and microvilli in several cell types. Equally important is Rac1’s ability to
decrease cellular stiffness via the resorption of these cellular processes to ensure proper cell–cell contact. On antigen recognition and chemokine stimulation of lymphocytes, Rac1 promotes
dephosphorylation of the ERM family (ezrin/radixin/moesin), which is known to cross link actin filaments with the plasma membrane52. These cellular events diminish T-cell membrane rigidity
allowing the dynamic resorption of cellular processes and optimal subsequent cellular contact with APCs. Therefore, investigating the effect of genetic deficiency or pharmacological
inhibition of the Tiam1/Rac1 complex on antigen recall, it is expected to see a weakened antigenic recognition ability as evidenced by the dampened proliferation of cultured splenocytes from
MOG35–55 immunized mice that were treated with NSC23766 and EHT1864 _in vivo_. In conclusion, we have demonstrated that Tiam1/Rac1 signalling is required for the encephalitogenicity of CD4+
T cells, very likely through its regulation of RORγt-mediated _Il17_ transcription. Our work herein demonstrates that the Tiam1/Rac1 complex controls this cytokine transcriptionally, but
more investigation will be required to further characterize the molecular details of this regulation. Moreover, the question of whether Tiam1/Rac1 signalling in T cells influences the
pathogenesis of other autoimmune or infectious diseases is intriguing. METHODS ANIMALS AND EAE INDUCTION WITH MOG35–55 Six- to eight-week-old female WT C57BL/6 mice were purchased from The
Jackson Laboratory. _Tiam1−_/− (Knockout) mice27 were provided by John G. Collard (Divisions of Cell Biology, The Netherlands Cancer Institute, Amsterdam, The Netherlands). The Tiam1
knockout mice were backcrossed repeatedly (8 × ) to FVB mice in the lab of origin (Collard lab). Subsequently, the mice has been backcrossed with C57BL/6 for 8 generations in our lab.
C57BL/6, _Rac1_fl/fl (ref. 53) (on a mixed 129S4/SvJae, BALB/c, C57BL/6 background), and _Rorc_fl/fl mice54 (on a mixed C57BL/6J:C67/BL6N genetic background) were purchased from The Jackson
Laboratory and were bred with _CD4-Cre_ transgenic mice (Taconic) for obtaining T-cell-specific Rac1-Rac1-null and RORγ-RORγt-null mice, respectively. STAT3 floxed mice (on C57BL/6
background) were previously described55 and were crossed with _CD4-Cre_ transgenic mice. EAE was induced by subcutaneously immunizing mice in the flanks with myelin oligodendrocyte
glycoprotein peptide (MOG)35–55 (New England Peptide). The 100 μg of administered (MOG)35–55 was comprised of 50 μl PBS and 50 μl CFA containing 250 ng _Myobacterium tuberculosis_ (Fisher
Scientific). Intraperitoneal injection of 200 ng pertussis toxin (List Biological Laboratories) followed on the day of immunization as well as 2 days later. ADOPTIVE TRANSFER AND TREATMENT
WITH TIAM1/RAC1 INHIBITORS For the adoptive transfer of _Tiam1−_/− T cells, _Tiam1−_/− and _Tiam1+_/+ mice were immunized with MOG35–55/CFA for 8 days, draining LN cells were isolated and
re-activated with MOG35–55 peptides (20 μg) in the presence of mouse recombinant IL-23 (20 ng ml−1) for 2 days, CD4+ T cells were isolated by MACS magnetic beads and 0.75 million cells were
injected i.p. in Rag1 deficient mice. Recipient mice received PT injections (69 ng) on days 1 and 3 after cell transfer. For Rac1 pharmacological inhibition, WT mice received 5 mg kg−1 of
NSC23766 (Calbiochem), 40 mg kg−1 of EHT 1864 (TOCRIS) or PBS on the day of the immunization and then daily for 7 days. Animals were kept for observation and scoring for 30 days. EAE
clinical disease was scored accordingly: score 1, limp tail or isolated gait weakness without limp tail; score 2, gait weakness or partial hind and/or front limb paralysis; score 3, total
hind limb paralysis; score 4, total hind limb and partial front limb paralysis; score 5, moribund animal or death. Mice were housed in the New Research Building Animal Facility at Harvard
Medical School under specific pathogen-free conditions. All experiments involving animals were done with the approval of the Harvard Medical Area Standing Committee on Animals and in
compliance with their protocols. ANTIBODIES AND REAGENTS All FACS antibodies (Abs) and blocking Abs were purchased from BD Biosciences or eBioscience. Mouse anti β-actin monoclonal antibody
(mAb), rabbit anti-Tiam1 antibody, and mouse anti-Rac1 antibody used for western blot were purchased from Sigma-Aldrich, Millipore and BD Biosciences, respectively. Mouse monoclonal Tiam1
antibody from Santa Cruz Biotechnology was used for immunoprecipitation. Other antibodies used for co-IP include mouse anti RORγ (BD Biosciences), mouse anti-Rac1 (Abcam), mouse anti lamin
A/C (Cell Signaling) and goat anti paxillin (Santa Cruz Biotechnology). For the proximal ligation assay, rabbit anti-Tiam1 (Santa Cruz Biotechnology), mouse anti-Rac1 (Abcam), mouse anti
RORγ (BD Biosciences) were used. All recombinant cytokines were purchased from R&D Systems. MOUSE AND HUMAN T-CELL DIFFERENTIATION Naive CD4+ T cells were purified from naive C57BL/6
mice using magnetic-activated cell sorting (MACS) (Miltenyi). Cells (250–500 × 103) were stimulated using plate-bound anti-CD3 (4 μg ml−1) as well as soluble anti-CD28 (2 μg ml−1) in 48-well
plates for 4 days in a serum-free medium (X-VIVO-20; Lonza) supplemented with 50 μM 2-mercaptoethanol, 1 mM sodium pyruvate, L-glutamine, nonessential amino acids, and 100 U ml−1 of
penicillin and streptomycin in the presence of recombinant cytokines. Naive CD4+ T cells were polarized in the presence of recombinant mouse IL-12 (10 ng ml−1) and anti-IL-4 neutralizing
antibody (10 μg ml−1) for Th1, recombinant mouse IL-4 (10 ng ml−1) plus anti-IFNγ antibody (10 μg ml−1) for Th2, recombinant mouse IL-6 (30 ng ml−1) and recombinant human TGF-β1 (3 ng ml−1)
plus anti-IFNγ (10 μg ml−1) for Th17. For pathogenic Th17 cells, recombinant IL-1β and IL-23 were used at 20 ng ml−1 when cells were polarized using IL-6, IL-1β and IL-23 during 4 days of
culture. In addition, pathogenic Th17 cells were alternatively polarized with IL-23 at 10 ng ml−1 for 48 h in the presence of IL-6 and TGF- β1 used to restimulate differentiated Th17 cells
that were rested prior for 2 days in the presence of IL-2 (2 ng ml−1). No recombinant cytokines or blocking Abs were added for Th0 condition. NSC23766 (94 μM) was added when indicated. For
human T-cell studies, blood was drawn from healthy control volunteers in compliance with the Institutional Review Board protocols. Ficoll-Paque PLUS (GE Healthcare) was used to separate
PBMCs by gradient centrifugation. Naive CD4+ T cells were subsequently isolated from fresh PBMCs by negative selection with beads using the CD4+ T-cell isolation kit II (Miltenyi). Cells
were cultured at 104 cells per well in serum-free X-Vivo 15 media (Lonza), and stimulated for 7 days with plate-bound anti-CD3 (UCHT1, 5 μg ml−1) and soluble anti-CD28 (28.2, 1 μg ml−1) Abs
in the presence of anti-IFN-γ plus anti-IL-4 for Th0, IL-12 (10 ng ml−1) plus anti-IL-4 for T helper 1, or TGF-β1 (5 ng ml−1), IL-6, IL-21, IL-23 (all at 25 ng ml−1) and IL-1β (12.5 ng ml−1)
for T helper 17 cells. Neutralizing Abs were used at 10 μg ml−1. All recombinant proteins were purchased from R&D Systems. Neutralizing antibodies were purchased from BD Biosciences.
FLOW CYTOMETRY For intracellular cytokine staining, _in vitro_ polarized cells or cells isolated from mice LNs, spleen or spinal cords were washed then stimulated in culture medium with
phorbol 12-myristate 13-acetate (PMA) (50 ng ml−1; Sigma-Aldrich) and ionomycin (300 ng ml−1; Sigma-Aldrich) for 4 h in the presence of GolgiStop (BD Biosciences) at 37 °C, in a humidified
5% CO2 chamber. The cells were then washed and stained for 20 min with 7AAD for dead cells exclusion and fluorochrome-labelled mAbs against surface cell markers. The cells were fixed and
permeabilized using Cytofix/Cytoperm (BD Biosciences) and Perm/Wash buffer (BD Biosciences). Following permeabilization, cells were stained with intracellular monoclonal antibodies 30–40 min
at 4 °C, washed with wash buffer, acquired on the LSR II (BD Biosciences), and analyzed using FlowJo software. For intracellular cytokine staining of T cells infiltrating the CNS, mice were
sacrificed and perfused with 20 ml of PBS. Spinal cord tissues were mechanically homogenized, resuspended in 30% Percoll, and slowly layered on top of 70% Percoll. After centrifugation at
1,300_g_ for 20 min, the inflammatory cells infiltrating the CNS were retrieved from the 30/70% Percoll interface. Lymphocytes were prepared for intracellular flow cytometry staining as
described above using the indicated antibodies. LUMINEX ASSAY Released cytokines expression was measured by fluorescent bead-based multiplex assay (Millipore) in accordance with the
manufacturer’s instructions. Samples were acquired with Luminex microplate reader and quantified using Upstate BeadView software. ELISPOT ASSAY Immobilon-P (PVDF) plates (Millipore, Bedford,
MA, USA) were activated using 35% ethanol for 1 min and coated with 4 μg ml−1 of purified mouse anti-IL-17A and anti-IFN-γ (both from BD Biosciences, San Diego, CA, USA) overnight at 4 °C.
Plates were blocked with 1% BSA, equal number of LN or spleen cells were loaded per well and incubated with increasing doses of MOG35–55 for 36 h at 37 °C. Plates were then washed and
biotinylated anti-IFN-γ and anti-IL-17A (2 μg ml−1) were added and incubated overnight at 4 °C. Afterwards, the plates were again washed and incubated at room temperature for 2 h with
alkaline phosphatase (Sigma, St Louis, MO, USA). Plates were then developed in BCIP/NBT (Sigma) solution. A computer-assisted ELISPOT Image Analyzer (Cellular Technology Limited, Cleveland,
OH, USA) was used to count the spots. PROLIFERATION ASSAY Cells were cultured in 96-well plate in triplicate at 2–5 × 105 cells per well, and 200 μl per well was plated with different
concentrations of MOG35–55 peptide. After 48 h of culture, 1 μCi 3H-thymidine (NEN Life Science Products, Boston, MA, USA) was added in 20 μl of media to each well for another 16–18 h. The
CPM per well was counted by scintillation counter (Perkin-Elmer, Waltham, MA, USA). Data is presented as the mean CPM of triplicate wells per condition. WESTERN BLOTTING Cells were lysed in
RIPA buffer (25 mM Tris·HCl pH 7.6, 150 mM NaCl, 1% NP40, 1% sodium deoxycholate, 0.1% SDS, Thermo Scientific) used alongside protease and phosphatase inhibitor mixtures (Roche Diagnostics
and Sigma-Aldrich, respectively). For separation by electrophoresis, 20 μg of total protein was loaded onto a SDS–PAGE gel according to standard protocols and then transferred on a PVDF
membrane. The resulting membranes were then blocked for 1 h using 5% powder skim milk in TBS-Tween 20 and afterwards were incubated overnight at 4 °C with primary Abs including Tiam1 rabbit
mAb (Millipore, ST1070, dilution 1/1,000) and Rac1 rabbit mAb (BD Biosciences, Cat. No. 610650, dilution 1/1,000. Membranes were striped and re-probed with mouse anti-β-actin antibody
(Sigma-Aldrich, A3854, dilution 1/500,000) as a loading control. Blots were washed 3–5 times with TBS-Tween 20 and then incubated for 1 h using the appropriate HRP-conjugated secondary
antibodies. Lastly, membranes were developed with the Immobilon Western Chemiluminescent HRP kit (Millipore, Billerica, MA, USA). Western blot images included in Fig. 1b have been cropped
for presentation. Full size images of western blots are presented in Supplementary Fig. 1. PROXIMITY LIGATION ASSAY (PLA) All antibodies used in this assay (rabbit anti-Tiam1 (Santa Cruz
Biotechnology, sc-872), mouse anti-Rac1 (abcam, ab33186), mouse anti RORγ (BD Biosciences, Cat. No. 562663)) were antecedently rigorously tested with western blot and
immunocytochemistry/immunofluorescence. The specificity of the antibodies was assessed by the time course of expression and the subcellular localization of the signal. PLA was performed
according to the manufacturers protocol (Duolink In Situ Red Starter Kit Mouse/Rabbit, DUO92101, Sigma), with minor modifications. Briefly, naive CD4 T cells were subjected to Th17
differentiation conditions. Cells were harvested at 0, 6, 12, 24 and 48 h after the initiation of differentiation, fixed with 4% paraformaldehyde (PFA) in PBS for 10 min at room temperature.
Subsequently the cells were washed three times with PBS (5 min each wash) and blocked with blocking buffer (PBS containing 8% horse serum, 3% bovine serum albumin and 0.3% Triton-X) for 30
min at room temperature. At this point cells were divided into two batches, on which two parallel PLA assays were performed: one for the detection of Tiam/Rac1 interaction and one for the
detection of Tiam1/RORγ interaction. The primary antibodies (rabbit anti-Tiam1 with mouse anti-Rac1 for one of the batches, and rabbit anti-Tiam1 with mouse anti RORγ for the other batch)
were applied on the cells in blocking buffer overnight at 4 °C on an orbital shaker. On the following day, cells were washed three times with PBS (5 min each wash) and incubated with the ‘+’
and the ‘−’ PLA probes diluted at 1:5 in the blocking buffer for 1 h at 37 °C with gentle shaking. Subsequently the cells were washed two times (5 min each) with 1 × Wash Buffer A, which
was provided with the kit. The cells were then incubated with the ligase containing ligation solution for 30 min at 37 °C with gentle shaking. Next, the cells were washed twice (2 min each)
with 1 × Wash Buffer A and then incubated with the polymerase containing amplification solution for 100 min at 37 °C with gentle shaking. Following the amplification, the cells were washed
two times (10 min each) with 1 × Wash Buffer B followed a 1 min was with 0.01 × Wash Buffer B. Cells were subsequently resuspended in a DAPI (nuclear counterstain) containing mounting medium
(supplied with the kit), mounted on a glass microscopy slide, covered with coverslip, sealed with nail polish and imaged as described below. CONFOCAL IMAGING AND IMAGE ANALYSIS A Zeiss
LSM710 confocal laser scanning microscope was used to acquire confocal images using the 405 and the 561 nm laser lines with the pinholes set to 1 Airy units for both channels. A 40 × oil
immersion objective was used (Zeiss EC-PlanNEOFLUAR 40 × /1.30 Oil DiC M27) with a 10 × digital zoom available in the Zeiss Zen acquisition software. The dimensions of the acquired 8 bit
images were X:256, Y:256 and Z:24 pixels, while the resolution was X:0.083 μm, Y:0.083 μm and Z:0.052 μm. The images were saved as.czi files and analyzed in FIJI (ImageJ2). The ‘3D object
counter’ function of FIJI (ImageJ2) was used to determine the number of Tiam1/Rac1 and Tiam1/RORγ interactions per cells. Due to the more than 500-fold signal amplification properties of the
PLA assay, each interaction of a single Tiam1 and Rac1 molecule (or Tiam1 and RORγ molecule) can be detected under the microscope as a fluorescent sphere. The nuclear counterstain (DAPI)
was used for image segmentation when determining the subcellular localization (cytoplasmic versus nuclear) of the PLA signal. The Z stacks of 10 cells were analyzed per time point for both
the Tiam1/Rac1 and the Tiam1/RORγ PLA assays. SUBCELLULAR FRACTIONATION AND CO-IP Naïve CD4 T cells were differentiated to Th17 cells for 48 h (as described above) and subsequently
fractionated using the NE-PER Nuclear and Cytoplasmic Extraction Kit (Thermo Scientific, Cat. No. 78833) according to the manufacturer’s instructions. Briefly, 4–5 million differentiated
CD4+ T cells were lysed in CER I, the lysate was vortexed, CER II was added to the lysate, vortexed again and let to sit on ice for 1 min. After centrifugation with 16,000_g_ for 5 min the
supernatant (the cytoplasmic fraction) was transferred to a clean tube. The pellet (containing the nuclei) was resuspended in ice-cold NER buffer, vortexed every 10 min for 40 min, and
subsequently spun down with 16,000_g_ for 10 min. The resulting supernatant (the nuclear fraction) was transferred to a pre-chilled microcentrifuge tube and the amounts were determined with
the Pierce BCA Kit (Thermo Scientific, Cat. No. 23225) according to the manufacturer’s instructions. For co-IP 100 μg of cytoplasmic protein and 100 μg of nuclear protein was used as input.
As a pre-clearing step the different fractions were pre-incubated with 125 μl of Protein A Magnetic Beads (New England Biolabs, S1425S) for 1 h on a rotational mixer at 4 °C. Subsequently
magnetic field was applied to the tubes, the supernatants transferred to clean tubes and the magnetic beads discarded. Each sample was incubated overnight with either 2 μg of rabbit
anti-Tiam1 antibody (Santa Cruz Biotechnology, sc-872) or 2 μg of rabbit IgG in a final volume of 1 ml on a rotating mixer at 4 °C. Next day 125 μl of Protein A Magnetic Beads per sample was
added to the cell lysates and incubated for 1 h on a rotational mixer at 4 °C. Subsequently, the magnetic beads were separated from the lysates with a magnet and washed three times with IP
buffer (Thermo Scientific, Cat. No. 87788). Finally, the co-immunoprecipitated proteins were eluted from the beads with loading buffer (containing LDS Sample Buffer (Invitrogen, NP0007) and
Sample Reducing Agent (Invitrogen, NP0009), loaded on a 4–12% Bis-Tris gel and separated by electrophoresis. The immunoprecipitated and co-immunoprecipitated proteins were detected by the
following antibodies using the ECL detection method (Thermo Scientific, Cat. No. 32109): mouse anti RORγ (BD Biosciences, Cat. No. 562663, dilution 1/500), mouse anti-Rac1 (Abcam, ab33186,
dilution 1/500), mouse anti lamin A/C (Cell Signaling, Cat. No. 4777, dilution 1/1,000) and goat anti paxillin (Santa Cruz Biotechnology, sc-7336, dilution 1/1,000). Immunoblot images
included in Fig. 5g have been cropped for presentation. Full size images of western blots are presented in Supplementary Fig. 7. EXPRESSION ANALYSIS BY REAL-TIME PCR RNA from 50–100 × 103 T
cells was purified using Stratagene RNA kit and directly transferred into the RT reagent using the Applied Biosystems Taqman reverse transcriptase reagents. Samples were then subjected to
real-time PCR analysis on the Applied Biosystems PRISM 7000 Sequencer Detection System (Applied Biosystems, Foster City, CA, USA) using standard conditions. Genes analyzed were detected with
commercially available assays (Applied Biosystems). The relative mRNA abundance was normalized against GAPDH. CHIP-QPCR Naive CD4+ CD44lowCD62Lhi T cells were sorted by flow cell sorter and
were polarized to Th17 phenotype for 48 h. ChIP was performed according to the protocol described in Bassil _et al_.33 Cell lysates were used for immunoprecipitation with anti-RORγt,
anti-Tiam1, and anti-Rac1 (Santa Cruz Biotechnology: sc-28559, sc-872 and sc-217, respectively) and were compared with control IgG. For the CNS2 ChIP, the following primers were used for the
detection of RORγt, Tiam1 and Rac1 binding: Fwd: ATGGGCCTCTCTTTCCACTGATG, Rev: GGAATTTGTGGTGGAAGGGAGTG according to previous report36. LUCIFERASE REPORTER ASSAY PGL3 luciferase reporter
plasmids encoding the _Il17_ promoter with CNS2 and control plasmid were kindly provided by Dr Chen Dong36. Human embryonic kidney 293T cells were seeded in culture plates the day before
transfection according the manufacturer’s instructions. The cells confluency at the day of the transfection was 40–80%. 293T cells were transfected with luciferase reporter constructs
together with the indicated plasmids using Effectene Transfection Reagent kit (Qiagen). Cells were cultured overnight then treated with Luciferase Assay System kit reagents (Promega), and
results were acquired on Wallac 1420 Viktor2 plate reader (Perkin-Elmer). HISTOLOGICAL ANALYSIS Fourteen days after immunization, mice were sacrificed, and spinal cords were fixed in Bouin’s
fixative and then embedded in paraffin. Slides were processed for hematoxylin and eosin stains, and foci of inflammation were counted in a blinded fashion. STATISTICAL ANALYSIS The
Mann–Whitney test was used for analysis of clinical disease. Statistical evaluations of luciferase activity, cytokine production and cell frequency measurements were performed using the
unpaired Student’s _t_-test. Values of _P_<0.05 were considered to be statistically significant. DATA AVAILABILITY Microarray data of CD4+ T cells from MS patients and healthy controls
was downloaded from Gene Expression Omnibus (accession code GSE32988). The authors declare that the data that supporting the findings of this study are available within the article and its
Supplementary Information Files. ADDITIONAL INFORMATION HOW TO CITE THIS ARTICLE: Kurdi, A. T. _et al_. Tiam1/Rac1 complex controls _Il17a_ transcription and autoimmunity. _Nat. Commun._ 7,
13048 doi: 10.1038/ncomms13048 (2016). REFERENCES * Brucklacher-Waldert, V. et al. Phenotypical and functional characterization of T helper 17 cells in multiple sclerosis. _Brain_ 132,
3329–3341 (2009). Article Google Scholar * Yang, X. O. et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. _Immunity_ 28, 29–39
(2008). Article CAS Google Scholar * Bettelli, E. et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. _Nature_ 441, 235–238
(2006). Article ADS CAS Google Scholar * Miossec, P., Korn, T. & Kuchroo, V. K. Interleukin-17 and type 17 helper T cells. _N. Engl. J. Med._ 361, 888–898 (2009). Article CAS
Google Scholar * Miossec, P. & Kolls, J. K. Targeting IL-17 and TH17 cells in chronic inflammation. _Nat. Rev. Drug Discov._ 11, 763–776 (2012). Article CAS Google Scholar *
Komiyama, Y. et al. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. _J. Immunol._ 177, 566–573 (2006). Article CAS Google Scholar * Hueber,
W. et al. Effects of AIN457, a fully human antibody to interleukin-17A, on psoriasis, rheumatoid arthritis, and uveitis. _Sci. Transl. Med._ 2, 52ra72 (2010). Article Google Scholar *
Deiss, A. et al. Treating multiple sclerosis with monoclonal antibodies: a 2013 update. _Expert Rev. Neurother._ 13, 313–335 (2013). Article CAS Google Scholar * Patel, D. D. et al.
Effect of IL-17A blockade with secukinumab in autoimmune diseases. _Ann. Rheum. Dis._ 72, (Suppl 2): ii116–ii123 (2013). Article CAS Google Scholar * Genovese, M. C. et al. LY2439821, a
humanized anti-interleukin-17 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: a phase I randomized, double-blind, placebo-controlled, proof-of-concept study.
_Arthritis Rheum._ 62, 929–939 (2010). Article CAS Google Scholar * Leonardi, C. et al. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. _N. Engl. J. Med._
366, 1190–1199 (2012). Article CAS Google Scholar * Papp, K. A. et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. _N. Engl. J. Med._ 366, 1181–1189 (2012).
Article CAS Google Scholar * Ivanov, I. I. et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. _Cell_ 126, 1121–1133
(2006). Article CAS Google Scholar * Manel, N., Unutmaz, D. & Littman, D. R. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the
nuclear receptor RORgammat. _Nat. Immunol._ 9, 641–649 (2008). Article CAS Google Scholar * Lee, Y. et al. Induction and molecular signature of pathogenic TH17 cells. _Nat. Immunol._ 13,
991–999 (2012). Article CAS Google Scholar * McGeachy, M. J. et al. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology.
_Nat. Immunol._ 8, 1390–1397 (2007). Article CAS Google Scholar * McGeachy, M. J. & Cua, D. J. The link between IL-23 and Th17 cell-mediated immune pathologies. _Semin. Immunol._ 19,
372–376 (2007). Article CAS Google Scholar * Huh, J. R. & Littman, D. R. Small molecule inhibitors of RORgammat: targeting Th17 cells and other applications. _Eur. J. Immunol._ 42,
2232–2237 (2012). Article CAS Google Scholar * Yosef, N. et al. Dynamic regulatory network controlling TH17 cell differentiation. _Nature_ 496, 461–468 (2013). Article ADS CAS Google
Scholar * Purwar, R. et al. Robust tumor immunity to melanoma mediated by interleukin-9-producing T cells. _Nat. Med._ 18, 1248–1253 (2012). Article CAS Google Scholar * Xiao, S. et al.
Small-molecule RORgammat antagonists inhibit T helper 17 cell transcriptional network by divergent mechanisms. _Immunity_ 40, 477–489 (2014). Article CAS Google Scholar * Govek, E. E.,
Newey, S. E. & Van Aelst, L. The role of the Rho GTPases in neuronal development. _Genes Dev._ 19, 1–49 (2005). Article CAS Google Scholar * Van Aelst, L. & D'Souza-Schorey,
C. Rho GTPases and signaling networks. _Genes Dev._ 11, 2295–2322 (1997). Article CAS Google Scholar * Boissier, P. & Huynh-Do, U. The guanine nucleotide exchange factor Tiam1: a
Janus-faced molecule in cellular signaling. _Cell Signal_ 26, 483–491 (2014). Article CAS Google Scholar * Bid, H. K. et al. RAC1: an emerging therapeutic option for targeting cancer
angiogenesis and metastasis. _Mol. Cancer Ther._ 12, 1925–1934 (2013). Article CAS Google Scholar * Jamieson, C. et al. Rac1 augments Wnt signaling by stimulating beta-catenin-lymphoid
enhancer factor-1 complex assembly independent of beta-catenin nuclear import. _J. Cell Sci._ 128, 3933–3946 (2015). Article CAS Google Scholar * Malliri, A. et al. Mice deficient in the
Rac activator Tiam1 are resistant to Ras-induced skin tumours. _Nature_ 417, 867–871 (2002). Article ADS CAS Google Scholar * Arkin, M. Protein–protein interactions and cancer: small
molecules going in for the kill. _Curr. Opin. Chem. Biol._ 9, 317–324 (2005). Article CAS Google Scholar * Stebel, A. et al. Progression of breast tumors is accompanied by a decrease in
expression of the Rho guanine exchange factor Tiam1. _Oncol. Rep._ 21, 217–222 (2009). CAS PubMed Google Scholar * Hofbauer, S. W. et al. Tiam1/Rac1 signals contribute to the
proliferation and chemoresistance, but not motility, of chronic lymphocytic leukemia cells. _Blood_ 123, 2181–2188 (2014). Article CAS Google Scholar * Buongiorno, P. et al. Rac1 GTPase
and the Rac1 exchange factor Tiam1 associate with Wnt-responsive promoters to enhance beta-catenin/TCF-dependent transcription in colorectal cancer cells. _Mol. Cancer_ 7, 73 (2008). Article
Google Scholar * Yang, X. P. et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. _Nat. Immunol._ 12, 247–254 (2011). Article CAS
Google Scholar * Bassil, R. et al. BCL6 controls Th9 cell development by repressing Il9 transcription. _J. Immunol._ 193, 198–207 (2014). Article CAS Google Scholar * Gao, Y. et al.
Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. _Proc. Natl Acad. Sci. USA_ 101, 7618–7623 (2004). Article ADS CAS Google Scholar * Shutes, A. et
al. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. _J. Biol. Chem._ 282, 35666–35678 (2007). Article CAS Google Scholar *
Wang, X. et al. Transcription of Il17 and Il17f is controlled by conserved noncoding sequence 2. _Immunity_ 36, 23–31 (2012). Article Google Scholar * Michiels, F. et al. A role for Rac in
Tiam1-induced membrane ruffling and invasion. _Nature_ 375, 338–340 (1995). Article ADS CAS Google Scholar * Russell, S. E. et al. Soluble IL-2Ralpha (sCD25) exacerbates autoimmunity
and enhances the development of Th17 responses in mice. _PLoS ONE_ 7, e47748 (2012). Article ADS CAS Google Scholar * de Paula, A. S. A. et al. Autologous haematopoietic stem cell
transplantation reduces abnormalities in the expression of immune genes in multiple sclerosis. _Clin. Sci. (Lond.)_ 128, 111–120 (2015). Article Google Scholar * Michiels, F. et al.
Regulated membrane localization of Tiam1, mediated by the NH2-terminal pleckstrin homology domain, is required for Rac-dependent membrane ruffling and C-Jun NH2-terminal kinase activation.
_J. Cell Biol._ 137, 387–398 (1997). Article CAS Google Scholar * Michaelson, D. et al. Rac1 accumulates in the nucleus during the G2 phase of the cell cycle and promotes cell division.
_J. Cell Biol._ 181, 485–496 (2008). Article CAS Google Scholar * Chook, Y. M. & Blobel, G. Karyopherins and nuclear import. _Curr. Opin. Struct. Biol._ 11, 703–715 (2001). Article
CAS Google Scholar * Bourguignon, L. Y. et al. CD44 interaction with tiam1 promotes Rac1 signaling and hyaluronic acid-mediated breast tumor cell migration. _J. Biol. Chem._ 275, 1829–1838
(2000). Article CAS Google Scholar * Buchsbaum, R. J., Connolly, B. A. & Feig, L. A. Regulation of p70 S6 kinase by complex formation between the Rac guanine nucleotide exchange
factor (Rac-GEF) Tiam1 and the scaffold spinophilin. _J. Biol. Chem._ 278, 18833–18841 (2003). Article CAS Google Scholar * Buchsbaum, R. J., Connolly, B. A. & Feig, L. A. Interaction
of Rac exchange factors Tiam1 and Ras-GRF1 with a scaffold for the p38 mitogen-activated protein kinase cascade. _Mol. Cell Biol._ 22, 4073–4085 (2002). Article CAS Google Scholar *
El-Behi, M. et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. _Nat. Immunol._ 12, 568–575 (2011). Article CAS Google
Scholar * Yang, X. O. et al. Regulation of inflammatory responses by IL-17F. _J. Exp. Med._ 205, 1063–1075 (2008). Article CAS Google Scholar * Guo, X. et al. Balanced Tiam1-rac1 and
RhoA drives proliferation and invasion of pancreatic cancer cells. _Mol. Cancer Res._ 11, 230–239 (2013). Article CAS Google Scholar * Chan, A. et al. The GTPase Rac regulates the
proliferation and invasion of fibroblast-like synoviocytes from rheumatoid arthritis patients. _Mol. Med._ 13, 297–304 (2007). Article ADS CAS Google Scholar * Ma, J. et al. Role of
activated Rac1/Cdc42 in mediating endothelial cell proliferation and tumor angiogenesis in breast cancer. _PLoS ONE_ 8, e66275 (2013). Article ADS CAS Google Scholar * Rougerie, P. &
Delon, J. Rho GTPases: masters of T lymphocyte migration and activation. _Immunol. Lett._ 142, 1–13 (2012). Article CAS Google Scholar * Nijhara, R. et al. Rac1 mediates collapse of
microvilli on chemokine-activated T lymphocytes. _J. Immunol._ 173, 4985–4993 (2004). Article CAS Google Scholar * Glogauer, M. et al. Rac1 deletion in mouse neutrophils has selective
effects on neutrophil functions. _J. Immunol._ 170, 5652–5657 (2003). Article CAS Google Scholar * Tilley, S. L. et al. Retinoid-related orphan receptor gamma controls immunoglobulin
production and Th1/Th2 cytokine balance in the adaptive immune response to allergen. _J. Immunol._ 178, 3208–3218 (2007). Article CAS Google Scholar * Takatori, H. et al. Lymphoid tissue
inducer-like cells are an innate source of IL-17 and IL-22. _J. Exp. Med._ 206, 35–41 (2009). Article CAS Google Scholar Download references ACKNOWLEDGEMENTS We thank Dr John Collard (The
Netherlands Cancer Institute) for the generous gifts of C1199-Tiam1, and the Tiam1-null mice, and Dr Chen Dong (Anderson Cancer Center) for providing the CNS2-pGL3 reporter vectors. We also
thank Dr Jaime Imitola for discussion and comments on the manuscript. The research was supported by Awards from the National Institutes of Health (AI093838 and AI043496 to S.J.K.), the
Massachusetts Life Sciences Center (RG110219 to W.E.) and the National Multiple Sclerosis Society (RG3945 to S.J.K. and W.E., and PP1734 to W.E.). AUTHOR INFORMATION Author notes * Ahmed T.
Kurdi, Ribal Bassil and Marta Olah: These authors contributed equally to this work AUTHORS AND AFFILIATIONS * Ann Romney Center for Neurologic Diseases, Brigham and Women’s Hospital and
Harvard Medical School, Boston, 02115, Massachusetts, USA Ahmed T. Kurdi, Ribal Bassil, Chuan Wu, Sheng Xiao, Michael Frangieh, Thomas Buttrick, William Orent & Samia J. Khoury *
Department of Neurology, Program in Translational NeuroPsychiatric Genomics, Brigham and Women’s Hospital, Harvard Medical School, Broad Institute at Harvard University and MIT, Boston,
02115, Massachusetts, USA Marta Olah, Mariko Taga, Elizabeth M. Bradshaw & Wassim Elyaman * Abu Haidar Neuroscience Institute, American University of Beirut Medical Center, Riad El Solh,
1107 2020, Beirut, Lebanon Samia J. Khoury Authors * Ahmed T. Kurdi View author publications You can also search for this author inPubMed Google Scholar * Ribal Bassil View author
publications You can also search for this author inPubMed Google Scholar * Marta Olah View author publications You can also search for this author inPubMed Google Scholar * Chuan Wu View
author publications You can also search for this author inPubMed Google Scholar * Sheng Xiao View author publications You can also search for this author inPubMed Google Scholar * Mariko
Taga View author publications You can also search for this author inPubMed Google Scholar * Michael Frangieh View author publications You can also search for this author inPubMed Google
Scholar * Thomas Buttrick View author publications You can also search for this author inPubMed Google Scholar * William Orent View author publications You can also search for this author
inPubMed Google Scholar * Elizabeth M. Bradshaw View author publications You can also search for this author inPubMed Google Scholar * Samia J. Khoury View author publications You can also
search for this author inPubMed Google Scholar * Wassim Elyaman View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS A.T.K., R.B., M.O., C.W.,
S.X., M.F, T.B. and W.O. performed _in vitro_ and _in vivo_ experiments with murine cells; T.B. performed _in vitro_ experiments with human samples; E.M.B. designed human experiments; M.O.
performed bioinformatics analysis; A.T.K., R.B., M.O., S.J.K. and W.E. analyzed data and wrote the manuscript; S.J.K. and W.E. conceived and supervised the study and edited the manuscript.
CORRESPONDING AUTHORS Correspondence to Samia J. Khoury or Wassim Elyaman. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing financial interests. SUPPLEMENTARY
INFORMATION SUPPLEMENTARY INFORMATION Supplementary figures 1-9 (PDF 2280 kb) RIGHTS AND PERMISSIONS This work is licensed under a Creative Commons Attribution 4.0 International License. The
images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included
under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/ Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Kurdi, A., Bassil, R., Olah, M. _et al._ Tiam1/Rac1 complex controls _Il17a_
transcription and autoimmunity. _Nat Commun_ 7, 13048 (2016). https://doi.org/10.1038/ncomms13048 Download citation * Received: 07 April 2016 * Accepted: 30 August 2016 * Published: 11
October 2016 * DOI: https://doi.org/10.1038/ncomms13048 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable
link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative