C-Terminal Truncated Dystrophin Identified in Skeletal Muscle of an Asymptomatic Boy with a Novel Nonsense Mutation of the Dystrophin Gene
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

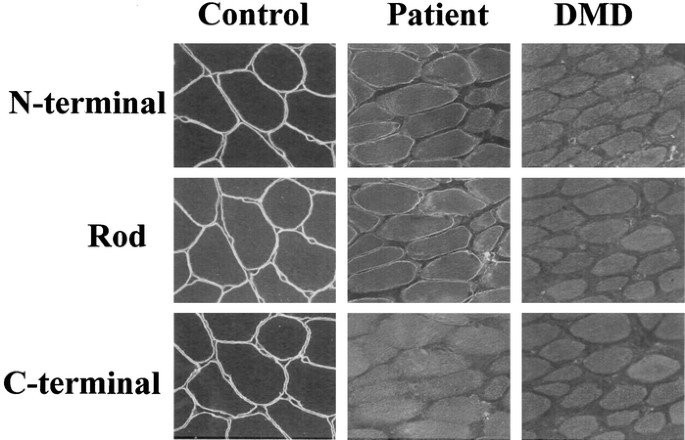
Mutations that cause premature stop codons in the dystrophin gene lead to a complete loss of dystrophin from skeletal muscle, resulting in severe Duchenne muscular dystrophy. Here, a
C-terminally truncated dystrophin resulting from a novel nonsense mutation is shown for the first time to be localized to the muscle plasma membrane. An asymptomatic 8-y-old boy was examined
for dystrophin in skeletal muscle because of high serum creatine kinase activity. Remarkably, no dystrophin labeling was seen with an MAb against the C-terminal domain, suggesting the
presence of an early stop codon in the dystrophin gene. Labeling with an antibody specific to the N-terminal domain, however, revealed weak, patchy, and discontinuous staining, suggesting
limited production of a truncated form of the protein. Molecular analysis revealed a novel nonsense mutation (Q3625X) as a result of a single nucleotide change in the patient's genomic DNA
(C10873T), leaving 1.6% of dystrophin gene product unsynthesized at the C terminus. Dystrophin mRNA analysis did not show rescue of the nonsense mutation as a result of exon-skipping by an
alternative splicing mechanism. This is the first report of an asymptomatic dystrophinopathy with a nonsense mutation in the dystrophin gene.
The severe Duchenne muscular dystrophy (DMD) and the more benign Becker muscular dystrophy (BMD) are allelic conditions characterized by progressive muscular degeneration and wasting
accompanied by an elevation of serum creatine kinase (CK). DMD is a rapidly progressive disease, with those affected starting to show muscle weakness at 4–5 y of age and losing the ability
to walk independently before the age of 12 y. BMD has a slower rate of progression; affected individuals remain ambulatory beyond the age of 16 y, and a few may lead near-normal lives (1).
DMD and BMD are caused by mutation of the dystrophin gene, which encodes a 14-kb mRNA that consists of 79 exons. The gene is the largest in humans and covers >3000 kb on the X chromosome
(2,3). DMD and BMD are the most common genetic muscle diseases, affecting >1 in 3,500 male births. Two thirds of DMD/BMD patients have deletion or duplication mutations of the dystrophin
gene, and their clinical progression can be predicted by whether the deletion or duplication maintains (in-frame) or disrupts (out-of-frame) the translational reading frame (the
reading-frame rule) (4). Dystrophin is absent from skeletal muscle of DMD, because the dystrophin that is produced is truncated as a result of the premature stop codon and therefore is
unstable, whereas in BMD, dystrophin that contains internal in-frame deletions produces protein that can be detected (5).
Single-base nonsense mutations have been suspected in DMD patients who do not show deletion/duplication mutations. However, detection of such defects in individual DMD patients is very
difficult as a result of the large size of the gene. More than 100 nonsense mutations have been reported at various points over a 14-kb length of the dystrophin mRNA (http://www.dmd.nl).
Despite this wide variation in coding potential (0–98.6% of the full-length protein), these truncating mutations are associated with a surprisingly uniform severity of the DMD phenotype (6).
However, a limited number of single-base nonsense mutations have been reported in patients with mild BMD that showed skipping of the exon encoding the mutation, thus producing an in-frame
mRNA (7–11).
Dystrophin is a cytoskeletal protein that is implicated in membrane stability and in communication between the extracellular matrix and the inner cytoskeleton (12,13). The protein, which
consists of 3685 amino acids, is divided into four distinct domains: an N-terminal domain, a large rod-like domain of 24 spectrin-like repeats that occupies >70% of its length, a
cysteine-rich domain, and, finally, a C-terminal domain (2,14). Studies conducted on DMD/BMD patients suggest that the N-terminal, cysteine-rich, and C-terminal domains are essential for
dystrophin's function (15,16). Notably, the C-terminal domain, which consists of 416 amino acids encoded by 13 exons, shows sequence similarity with only two other dystrophin-related
proteins and is considered to exert dystrophin's specific function (14,17,18). In fact, in-frame deletions that extend into the C-terminal domain have been reported to result in DMD, whereas
large in-frame deletions of the rod domain result in BMD (16).
Here we report a C-terminally truncated dystrophin caused by a mutation in an asymptomatic boy with high CK activity. We propose that nonsense mutations of the dystrophin gene can result in
a wide variety of clinical phenotypes.
The proband (KUDN 02765682) was an 8-y-old boy. His family history disclosed no neuromuscular disease. He started to walk independently at 1 y of age, and his motor development was normal.
He had a history of transient muscle weakness. At the age of 3 y, he complained of pain in the lower legs without any predisposing signs or symptoms and lost the ability to stand up and walk
by himself. His serum CK was found to be 4901 IU/L (normal