A highly D3R-selective and efficacious partial agonist (S)-ABS01-113 compared to its D3R-selective antagonist enantiomer (R)-ABS01-113 as potential treatments for opioid use disorder
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

The non-medical use of opioids has become a national crisis in the USA. Developing non-opioid pharmacotherapies for controlling this opioid epidemic is urgent. Dopamine D3 receptor (D3R)
antagonists and low efficacy partial agonists have shown promising profiles in animal models of opioid use disorders (OUD). However, to date, advancement to human studies has been limited.
Here we report the effects of (S)- and (R)-enantiomers of (±)-ABS01-113, structural analogs of the D3R partial agonist, (±)-VK4-40, in which the 3-OH in the linking chain is replaced by 3-F
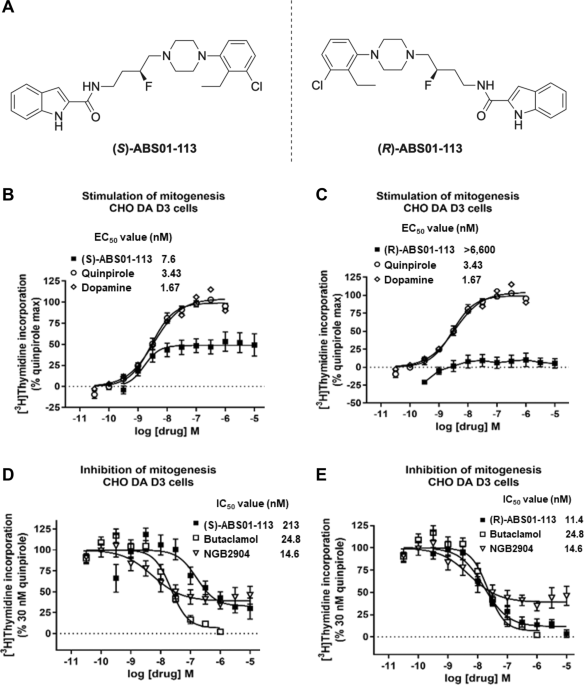
group. (S)- and (R)-ABS01-113 are identical in chemical structure but with opposite chirality. In vitro receptor binding and functional assays indicate that (S)-ABS01-113 is an efficacious
(55%) and potent (EC50 = 7.6 ± 3.9 nM) D3R partial agonist, while the (R)-enantiomer is a potent D3R antagonist (IC50 = 11.4 nM). Both (S)- and (R)-ABS01-113 bind with high affinity to D3R
(Ki = 0.84 ± 0.16 and 0.37 ± 0.06 nM, respectively); however, the (S)-enantiomer is more D3/D2-selective (>1000-fold). Pharmacokinetic analyses indicate that both enantiomers display
excellent oral bioavailability and high brain penetration. Systemic administration of (S)- or (R)-ABS01-113 alone failed to alter open-field locomotion in male rats and mice. Interestingly,
pretreatment with (S)- or (R)-ABS01-113 attenuated heroin-enhanced hyperactivity, heroin self-administration, and (heroin + cue)-induced reinstatement of drug-seeking behavior. Together,
these findings reveal that both enantiomers, particularly the highly selective and efficacious D3R partial agonist (S)-ABS01-113, demonstrate promising translational potential for the
treatment of OUD.
In the midst of the COVID-19 pandemic opioid overdose fatalities soared to a record 101,263 in 2021 [1]. This estimate translates to an average of 277 deaths/day. According to the CDC data,
more than 74% of the overdose deaths last year involved fentanyl and other opioids. Indeed, the rampant abuse of opioids continues to be a major public health problem. In addition, lockdowns
and social distancing made access to opioid-based treatments such as methadone, that require in person administration, much more difficult. Although the United States Food and Drug
Administration (FDA) has approved methadone and buprenorphine for the treatment of opioid use disorders (OUD), there is a high rate of relapse after cessation of treatment. Further concerns
for these opioid agonist therapies include their dependence liability and severe withdrawal symptoms after cessation of use [2]. Hence, developing non-opioid-based medications, without their
own dependence liability for the treatment of OUD, would provide a valuable alternative or addition to current pharmacotherapeutic options.
Over the past 2 decades, the dopamine D3 receptor (D3R) has become a target for medication development to treat substance use disorders and many D3R-selective ligands with high affinity have
been discovered [3,4,5,6]. In particular, selective D3R partial agonists and antagonists have been investigated extensively and have demonstrated promising results in experimental animals
[6,7,8,9,10,11]. However, to date, advancement to human studies has been limited to a very few selective D3R antagonists such as GSK598,809 [12, 13]. We previously focused on developing such
compounds as therapeutics to treat psychostimulant use disorders [3, 14, 15]. However, some concerns from the reports that D3R antagonists might exacerbate cardiotoxicity produced by
cocaine [16] led us to shift our attention to OUD, as the opioid crisis was pervasive and typically opioids do not produce cardiovascular toxicity [10, 17, 18]. Toward this goal, we used the
D3R crystal structure and extensive structure activity relationships (SAR) to develop bitopic D3R ligands [15, 19,20,21], which has led to the development of a number of highly selective
D3R antagonists or low efficacy partial agonists, exemplified by (±)-VK4-116 and (±)-VK4-40 [15, 22,23,24]. As these lead compounds have a chiral center in their linking chain (3-OH), the
synthesis and evaluation of the (S)- and (R)-enantiomers has continued [10, 23,24,25]. Extensive metabolism, pharmacokinetic (PK) and behavioral evaluation in numerous animal models of OUD
indicate both these lead molecules are effective in reducing opioid reward and relapse [6, 8, 10, 11] and further development toward human trials is underway.
We previously reported that replacing the 3-OH with a 3-F in the linking chain in these bitopic ligands resulted in some of the most selective D3R partial agonists/antagonists reported [26,
27]. Here, we report another novel pair of enantiomers — (S)- and (R)-ABS01-113, in which the 3-OH in the linking chain of (S)- or (R)-VK4-40 was replaced with a 3-F group [23]. Unlike the
low efficacy partial agonist (±)-VK4-40, (±)-ABS01-113 is a selective D3R antagonist, as is its (R)-enantiomer. However, we discovered that (S)-ABS01-113 is an efficacious D3R partial
agonist. As the only difference between these enantiomers is the linking chain chirality at the 3-position, rendering them mirror images of one another, we were curious to compare their
behavioral profiles and drug-like properties. We hypothesized that the (S)-enantiomer might not be as effective in these models as an antagonist or low efficacy partial agonist.
Nevertheless, we reasoned that if (S)-ABS01-113 produced a behavioral profile in our rodent models of OUD, it may have advantages over a D3R antagonist. Indeed, partial agonists have been
discovered to be more favorable pharmacotherapeutics for treatment of a variety of neuropsychiatric disorders ranging from OUD (e.g., buprenorphine) to schizophrenia (e.g., aripiprazole or
cariprazine) [28,29,30].
We had previously reported the synthesis and binding affinities at D3R and D2R of (S)- and (R)-ABS01-113 [20]. Herein we investigated their functional activity at D3R in vitro as well as
binding affinities at a few off-target sites with known homology to D3R. We also evaluated their brain: plasma ratios after intragastric drug administration. Lastly, we examined their
pharmacological efficacy in antagonizing opioid actions, including opioid-induced hyperactivity, self-administration, and reinstatement of drug-seeking behavior to predict their
translational potential for the treatment of OUD in humans.
(S)- and (R)-ABS01-113 off-target and functional activity assays were performed in Dr. Aaron Janowsky’s laboratory at the VA Portland Health Care System, Portland OR.
Mouse fibroblast cells expressing the human D1 receptor at high density (LhD1 cells) were used. The cells were grown to confluence in Dulbecco’s Minimal Essential Medium (DMEM) containing
10% FetalClone1 serum (FCS, HyClone), 0.05% penicillin-streptomycin (pen-strep), and 200 µg/ml of Geneticin (G418). One confluent 150 mm plate yielded enough membranes for 1 assay plate with
~10–15 µg protein/well. The cells from 1 150 mm plate were washed with phosphate-buffered saline (PBS), scraped into 50 mM Tris buffer (pH 7.4), and centrifuged at 27 K × g for 20 min. The
cell pellet was homogenized in 50 mM Tris (pH 7.4) with a polytron and centrifuged at 27 K × g for another 20 min. The pellet was overlaid with 2 ml of 50 mM Tris buffer (pH 7.4) and frozen
at −70 °C. On the day of experiment, each pellet was homogenized in 10 ml assay buffer (50 mM Tris-HCl, pH 7.4, containing 120 mM NaCl, 5 mM KCl, 2 mM CaCl2, and 1 mM MgCl2) with a polytron.
Cell homogenate (100 µl) was added to wells containing 800 µl of test drug (blinded to the investigator) or buffer. After 10 min preincubation, 100 µl of [3H]-SCH-23390 (0.18 nM final
concentration) was added. The plates were incubated at 25 °C for 60 min. The reaction was terminated by filtration through polyethylenimine-soaked (0.05%) filters using a Tomtec 96-well
harvester and radioactivity on the filters was counted using a Perkin Elmer microbetaplate 1405 liquid scintillation counter. Nonspecific binding was determined with 1 µM SCH-23390.
Human embryonic kidney cells coexpressing the human D4.4 receptor and adenylate cyclase type I (HEK-D4.4-AC1) were grown in DMEM supplemented with 5% FetalClone (HyClone), 5% bovine calf
serum (BCS), 0.05% pen-strep, 2 µg/ml of puromycin and 200 µg/ml of hygromycin. Membranes were prepared according to the procedures described for D1 cells, using 50 mM Tris (pH 7.4 at 4 °C).
Assay buffer contained 120 mM NaCl, 5 mM KCl, 1.5 mM CaCl2, 4 mM MgCl2, 1 mM ascorbic acid and 1 mM EDTA (pH 7.4 at 37 °C). One confluent 150 mm plate of D4.4 cells, yielded enough
membranes for 1 assay plate with ~14–25 µg protein/well. Proteins were determined using a BCA assay (Pierce). Cell homogenate (100 µl) was added to wells containing 800 µl of test drug
(blinded to the investigator) or buffer. After 10 min, 100 µl of [3H]-spiperone (specific activity 80.2, 0.2–0.3 nM final concentration) was added. The plates were incubated at 37 °C for 60
min. The reaction was terminated by filtration through polyethylenimine-soaked (0.05%) filters using a Tomtec 96-well harvester. Radioactivity on the filters was counted using a Perkin Elmer
microbeta counter. Nonspecific binding was determined with 1 µM haloperidol.
Human embryonic kidney cells expressing the human 5HT1A receptor (HEK-h5HT1A) were used. The cells were grown to confluence in DMEM containing 5% FetalClone (FC, HyClone), 5% bovine calf
serum, 0.05% penicillin-streptomycin (pen-strep), and 200 µg/mL of Geneticin (G418). The cells were scraped from 150 mm plates into phosphate-buffered saline and centrifuged at 1000 × g, for
10 min. The cell pellet was homogenized in 50 mM Tris-HCl (pH 7.4) with a polytron, and centrifuged at 27,000 × g. The homogenization and centrifugation were repeated twice to wash any
remaining 5-HT from the growth media. The final pellet was covered with 2 ml Tris buffer and stored at −80 °C until needed. The assay was performed in duplicate in a 96-well plate. Serial
dilutions of test compounds were made using the Biomek 4000 robotics system. The reaction mixture contained compound whose identity was not known to the experimenter, 100 µl of cell
homogenate (0.05 mg protein/well) and 100 µl of [3H]8-OH-DPAT (0.5 nM final concentration, 170 Ci/mmol, Perkin Elmer) and assay buffer (25 mM Tris-HCl, pH 7.4, containing 100 µM ascorbic
acid and 10 µM pargyline) in a final volume of 1 ml. Nonspecific binding was determined with 1.0 µM dihydroergotamine. The plates were incubated at room temperature for 60 min and then
filtered through polyethylenimine-soaked (0.05%) “A” filtermats on a Tomtec cell harvester. The filters were washed with cold 50 mM Tris buffer (pH 7.4) for 6 s, dried, spotted with
scintillation cocktail, and counted for 2 min, after a 4 h delay, on a Perkin Elmer microbetaplate counter. IC50 values were calculated with GraphPad Prism, and IC50 values were converted to
Ki values using the Cheng–Prusoff correction [31].
The method was adapted from AR Knight et al. [32]. Human embryonic kidney cells expressing the human 5HT2A receptor (HEK-h5HT2A) or human 5HT2C receptor (HEK-h5HT2C) were used. The cells
were grown until confluent on 15 cm plates. Media was removed, cells were washed with phosphate-buffered saline (PBS), scraped into 10 ml PBS and centrifuged at 1000 × g, for 10 min. The
pellet was resusupended in 10 ml, and polytronned at setting 6 for 5 s. The homogenate was centrifuged at 27,000 × g for 20 min. To minimize the residual 5-HT concentration, the pellet was
resuspended in buffer, polytronned, and centrifuged as above. The final pellet was resuspended in 2 ml buffer/plate of cells.
The binding assay included 50 µl drug, 5-HT or buffer, 50 µl cell homogenate, 50 µl [3H]5-HT (~4 nM) and assay buffer (50 mM Tris, pH 7.4 at 37 °C, with 0.1% ascorbic acid and 5 mM CaCl2)
buffer in a final volume of 250 µl. Specific binding was defined as the difference between total binding and binding in the presence of 10 µM 5-HT. The reaction was incubated for 1 h at 37
°C, and terminated by filtration through Wallac A filtermats presoaked in 0.05% polyethylenimine using a Tomtec 96-well harvester. Radioactivity remaining on filters was counted in a Perkin
Elmer microbetaplate counter. IC50 values were calculated using GraphPad Prism. IC50 values were converted to Ki values using the Cheng–Prusoff equation [31].
CHOp-D3 cells were maintained in alpha-MEM with 10% fetal bovine serum (FBS, Atlas Biologicals), 0.05% pen-strep, and 200 µg/ml of G418. To measure D3 stimulation of mitogenesis (agonist
assay) or inhibition of quinpirole stimulation of mitogenesis (antagonist assay), CHOp-D3 cells were seeded in a 96-well plate at a concentration of 5000 cells/well. The cells were incubated
at 37 °C in alpha-MEM with 10% FBS, pen-strep, and G418. After 48–72 h, the cells were rinsed twice with serum-free alpha-MEM and incubated for 24 h at 37 °C. Serial dilutions of test
compounds were made by the Biomek robotics system in serum-free alpha-MEM. In the functional assay for agonists, the medium was removed and replaced with 100 µl of test compound in
serum-free alpha-MEM. In the antagonist assay, the serial dilution of the putative antagonist test compound was added in 90 μl (1.1× of final concentration) and 300 nM quinpirole (30 nM
final) was added in 10 μl. After another 16-h incubation at 37 °C, 0.3 µCi of [3H]-thymidine in alpha-MEM supplemented with 10% FBS was added to each well and the plates were further
incubated for 2 h at 37 °C. The cells were trypsinized by addition of 10× trypsin solution (1% trypsin in calcium–magnesium-free phosphate-buffered saline) and the plates were filtered and
counted as usual. Quinpirole was run each day as an internal control and dopamine was included for comparative purposes.
To determine whether the enantiomers were orally available and brain penetrable, we evaluated plasma vs. brain drug concentrations after intragastric administration of (R)-ABS01-113 or
(S)-ABS01-113 in male Long–Evans rats (Charles River Laboratories, Frederick, MD) (initially weighing 250–300 g). The experimental procedures are the same as we reported before [10]. The
experimental methods in detail are provided in the Supplementary Materials.
Increased DA release produces an increase in open-field locomotion by activation of DA receptors [33, 34]. However, as D3R are not highly expressed in brain regions associated with motor
activity such as the dorsal striatum [3], we speculated that neither the D3R partial agonist (S)-ABS01-113 nor the D3R antagonist (R)-ABS01-113 would produce significant locomotor activity
by itself. Hence, we tested (S)- and (R)-ABS01-113 on locomotor activity in wild-type mice. The experimental methods in detail are provided in the Supplementary Materials.
Male Long–Evans rats (Charles River Laboratories, Frederick, MD) (initially weighing 250–300 g, n = 48) were used for heroin self-administration and reinstatement tests. Male wild-type
(C57/BL6J, 25–30 g) mice were used for open-field locomotion and sucrose self-administration tests. We chose males in this initial proof-of-concept study. However, females will be used in
our follow-up studies. Both rats and mice were chosen based on the availability of the test compounds, experimental animals, and equipment systems in the laboratory. To the best of our
knowledge, there have been no reports of sex differences in D3R antagonist or partial agonist behavioral effects. During heroin self-administration experimentation animals were housed
individually and mice used in the locomotor experiment were housed in groups of 4/cage in climate-controlled animal colony rooms on a reversed 12-h light–dark cycle (lights on at 7 a.m.)
with free access to food and water. All experiments were conducted during the animals’ active period (reversed dark cycle). The housing conditions and care of the animals were consistent
with the Guide for the Care and Use of Laboratory Animals. The protocols used in the present experiments were approved by the National Institute on Drug Abuse Animal Care and Use Committee.
To determine whether (S)- and (R)-ABS01-113 attenuated heroin intake, rats underwent intravenous catheter surgeries and intravenous self-administration procedures as described previously [9]
(see details in the Supplementary Materials).
The i.v. heroin self-administration procedures were the same as reported previously [10, 11]. Heroin self-administration training was conducted in an operant conditioning chamber equipped
with two response levers (Med Associates Inc., Georgia, VT, USA). Each rat was allowed to press the active lever for heroin (0.0125 or 0.05 mg/kg/infusion) under a FR1 schedule of
reinforcement during daily 3-h sessions. We used two heroin doses in order to determine whether the pharmacological efficacy of both the compounds is heroin dose dependent. In addition, both
the heroin doses are located on the descending limb of the heroin self-administration dose-response curve [34,35,36]. Using two different doses of heroin would help us to determine whether
the test compounds produce an increase or a decrease in heroin’s rewarding effects (or efficacy) based on the negative correlation between the number of heroin infusions and heroin dose —
higher numbers of heroin self-administration (infusions) at lower doses, while lower numbers of heroin self-administration at higher heroin doses. The experimental methods in detail are
provided in the Supplementary materials.
Procedures for oral sucrose self-administration in mice were the same as we reported previously [37] (see details in the Supplementary materials). After the active lever response was
stabilized, we observed the effects of the same doses of (R)- or (S)-ABS01-113 on oral sucrose self-administration.
We then examined whether (S)- or (R)-ABS01-113 can also reduce (heroin + cue)-induced reinstatement of drug seeking. A total of 57 rats with a history of heroin self-administration were used
in this experiment. After stable heroin self-administration was achieved, the animals underwent extinction training for 3 weeks. Each extinction session lasted for 3 h and responding on
either lever produced no consequences — no heroin or heroin-related cues. This phase continued until extinction criteria were met, defined as 0.05) although it revealed a significant time
main effect (F11,77 = 16.83, p 0.05). Given that (S)-ABS01-113, at 30 mg/kg, appeared to produce an increase in open-field locomotion beginning at 45 min after (S)-ABS01-113 administration
(Fig. 3A), we repeated this experiment. We found that (S)-ABS01-113, at the same drug doses, failed to alter locomotion within 2 h after the drug administration (Fig. S1).
A, B (S)- and (R)-ABS01-113 alone failed to alter open-field locomotion; C, D Pretreatment with (S)- or (R)-ABS01-113 decreased heroin-enhanced locomotor activity at lower doses (10 mg/kg).
Within-subjects design, n = 8 in each treatment. *p