Mining zebrafish microbiota reveals key community-level resistance against fish pathogen infection
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

The long-known resistance to pathogens provided by host-associated microbiota fostered the notion that adding protective bacteria could prevent or attenuate infection. However, the
identification of endogenous or exogenous bacteria conferring such protection is often hindered by the complexity of host microbial communities. Here, we used zebrafish and the fish pathogen
Flavobacterium columnare as a model system to study the determinants of microbiota-associated colonization resistance. We compared infection susceptibility in germ-free, conventional and
reconventionalized larvae and showed that a consortium of 10 culturable bacterial species are sufficient to protect zebrafish. Whereas survival to F. columnare infection does not rely on
host innate immunity, we used antibiotic dysbiosis to alter zebrafish microbiota composition, leading to the identification of two different protection strategies. We first identified that
the bacterium Chryseobacterium massiliae individually protects both larvae and adult zebrafish. We also showed that an assembly of 9 endogenous zebrafish species that do not otherwise
protect individually confer a community-level resistance to infection. Our study therefore provides a rational approach to identify key endogenous protecting bacteria and promising
candidates to engineer resilient microbial communities. It also shows how direct experimental analysis of colonization resistance in low-complexity in vivo models can reveal unsuspected
ecological strategies at play in microbiota-based protection against pathogens.
Animal resident microbial consortia form complex and long-term associations with important community-level functions essential for host development and physiology [1, 2]. Microbial
ecosystems also provide protection against exogenous pathogens by inhibition of pathogen settlement and growth and/or stimulation of the host immune system [3,4,5,6,7,8]. From the
perspective of microbial community composition, a shift or reduction in resident microbial diversity, a phenomenon generally referred to as dysbiosis, is often associated with increased
susceptibility to infection due to the loss or change in abundance of key microbial community members [3, 9]. These observations early supported the notion that addition or promotion of
individually or communally protective bacteria (such as probiotics) could minimize microbiota dysbiosis or directly prevent infection to restore host health [10,11,–12].
Although the efficacy of probiotics has been shown in animals and humans, their mechanisms of action are poorly understood and low throughput experimental models often offer limited
information on the individual contribution of probiotic species to community functions [1, 6, 7, 13, 14]. Moreover, characterization of bacterial strains improving colonization resistance is
still hindered by the complexity of host-commensal ecosystems. Zebrafish have recently emerged as a powerful tool to study microbe-microbe and host-microbe interactions [15,16,17,18,–19].
Zebrafish can be easily reared germ-free or gnotobiotically in association with specific bacterial species [15, 20]. Moreover, zebrafish bacterial communities are increasingly well
characterized and a number of phylogenetically distinct zebrafish gut bacteria can be cultured, making this model system directly amenable to microbiota manipulation and assessment of
probiotic effect on host infection resistance [21,22,23,–24]. Several studies have used zebrafish to evaluate the effect of exogenous addition of potential probiotics on host resistance to
infection by various pathogens [22,23,24,25,26,27,28,–29]. However, the role of the endogenous microbial community in protecting against invasive pathogens was rarely assessed and the
reported protections were often partial, illustrating the difficulty in identifying fully protective exogenous probiotics.
One major fish pathogen causing such problematic seasonal outbreaks is Flavobacterium columnare, a ubiquitously distributed freshwater bacterium that is the etiological agent of columnaris
disease [30]. This disease affects a broad range of wild and cultured species including carp, channel catfish, goldfish, eel, salmonids and tilapia [30,31,32,33,–34]. Different F. columnare
strains exhibit different degrees of virulence but relatively similar infection phenotypes [30, 31, 35,36,37]. The symptoms primarily associated with strains with low virulence are gross
tissue damages of gills, skin, fins, and tail, whilst such damages are not observed in highly virulent strains, leading to mortality within hours [31, 38]. Although F. columnare infection
causes important losses in aquaculture, there is no consensus on the determinants of its virulence. Recently, however, type IX secretion system (T9SS) was shown to be involved in F.
columnare pathogenesis in adult zebrafish, but the nature of the secreted virulence factors remains unclear [39].
Here we used germ-free and conventional zebrafish larvae to mine the indigenous commensal microbiota for bacterial species protecting against F. columnare. We identified two distinct
infection resistance strategies preventing mortality caused by F. columnare, mediated either by an individual member of the microbiota, the Bacteroidetes Chryseobacterium massiliae or by an
assembly of 9 individually non-protecting bacterial species that formed a protective community. Our results demonstrated that mining host microbiota constitutes a powerful approach to
identify key mediators of intrinsic colonization resistance, providing insight into how to engineer ecologically resilient and protective microbial communities.
Bacterial strains isolated from zebrafish microbiota are listed in Table 1. F. columnare strains (Supplementary Table S1) were grown at 28 °C in tryptone yeast extract salts (TYES) broth
[0.4% (w/v) tryptone, 0.04% yeast extract, 0.05% (w/v) MgSO4 7H2O, 0.02% (w/v) CaCl2 2H2O, 0.05% (w/v) D-glucose, pH 7.2]. F. columnare strains were assigned into four genomovar groups using
16S rRNA restriction fragment length polymorphism analysis, including genomovar I, I/II, II, and III [40]. All 10 strains of the core zebrafish microbiota species were grown in TYES or LB
at 28 °C.
Wild-type AB fish, originally purchased from the Zebrafish International Resource Center (Eugene, OR, USA), or myd88-null mutants (myd88hu3568/hu3568) [41], kindly provided by A.H. Meijer,
(Leiden University, the Netherlands), were raised in our facility. A few hours after spawning, eggs were collected, rinsed, and sorted under a dissecting scope to remove faeces and
unfertilized eggs. All following procedures were performed in a laminar microbiological cabinet with single-use disposable plasticware. Fish were kept in sterile 25 cm3 vented cap culture
flasks containing 20 mL of water (0-6 days post fertilization (dpf), 15 fish per flask) or 24-well microtiter plates (6-15 dpf,1 fish per 2 mL well) in autoclaved mineral water (Volvic) at
28 °C. Fish were fed 3 times a week from 4 dpf with germ-free Tetrahymena thermophila protozoans [22]. Germ-free zebrafish were produced after sterilizing the egg chorion protecting the
otherwise sterile egg, with antibiotic and chemical treatments (see below), whereas conventional larvae (with facility-innate microbiota) were directly reared from non-sterilized eggs and
then handled exactly as the germ-free larvae.
Egg sterilization was performed as previously described with some modifications [22]. Freshly fertilized zebrafish eggs were first bleached (0.003%) for 5 min, washed 3 times in sterile
water under gentle agitation and maintained overnight in groups of 100 eggs per 75 cm3 culture flasks with vented caps containing 100 mL of autoclaved Volvic mineral water supplemented with
methylene blue solution (0.3 µg/mL). Afterwards, eggs were transferred into 50 mL Falcon tubes (100 eggs per tube) and treated with a mixture of antibiotics (500 μL of penicillin G:
streptomycin, 10,000 U/ml: 10 mg/mL GIBCO #P4333), 200 μL of filtered kanamycin sulfate (100 mg/mL, SERVA Electrophoresis #26899) and antifungal drug (50 μL of amphotericin B solution
Sigma-Aldrich (250 μg/mL) #A2942) for 2 h under agitation at 28 °C. Eggs were then washed 3 times in sterile water under gentle agitation and bleached (0.003%) for 15 min, resuspending the
eggs every 3 min by inversion. Eggs were washed again 3 times in water and incubated 10 min with 0.01% Romeiod (COFA, Coopérative Française de l’Aquaculture). Finally, eggs were washed 3
times in water and transferred into 25 cm3 culture flasks with vented caps containing 20 mL of water. After sterilization, eggs were transferred with approximately 30 to 35 eggs / flasks and
were transferred into new flasks at 4 dpf before reconventionalization with 10 to 15 fish / flask. We monitored sterility at several points during the experiment by spotting 50 μL of water
from each flask on LB, TYES and on YPD agar plates, all incubated at 28 °C under aerobic conditions. Plates were left for at least 3 days to allow slow-growing organisms to multiply. Spot
checks for bacterial contamination were also carried out by PCR amplification of water samples with the 16S rRNA gene primers and procedure detailed further below. If a particular flask was
contaminated, those fish were removed from the experiment.
After hatching, fish were fed with germ-free T. thermophila 3 times per week from 4 dpf onwards. (i) T. thermophila stock. A germ-free line of T. thermophila was maintained at 28 °C in 20 mL
of PPYE (0.25% proteose peptone BD Bacto #211684, 0.25% yeast extract BD Bacto #212750) supplemented with penicillin G (10 unit/mL) and streptomycin (10 µg/mL). Medium was inoculated with
100 μL of the preceding T. thermophila stock. After one week of growth, samples were taken, tested for sterility on LB, TYES, and YPD plates and restocked again. (ii) Growth. T. thermophila
were incubated at 28 °C in MYE broth (1% milk powder, 1% yeast extract) inoculated from stock suspension at a 1:50 ratio. After 24 h of growth, T. thermophila were transferred to Falcon
tubes and washed (4400 rpm, 3 min at 25 °C) 3 times in 50 mL of autoclaved Volvic water. Finally, T. thermophila were resuspended in sterile water and added to culture flasks (500 µL in 20
mL) or 24-well plates (50 µL / well). Sterility of T. thermophila was tested by plating and 16S rRNA PCR as described in the section above.
(iii) Fine-powder feeding. When indicated, fish were fed with previously γ-ray-sterilized fine-powdered food suitable for an early first feeding gape size (ZM-000 fish feed, ZM Ltd) every 48
h [42].
At 4 dpf, just after hatching, zebrafish larvae were reconventionalized with a single bacterial population or a mix of several. The 10 bacterial strains constituting the core protective
microbiota were grown for 24 h in suitable media (TYES or LB) at 28 °C. Bacteria were then pelleted and washed twice in sterile water, and all adjusted to the same cell density (OD600 = 1 or
5.107 colony forming units (cfu)/mL) (i) Reconventionalization with individual species. Bacteria were resuspended and transferred to culture flasks containing germ-free fish at a final
concentration of 5.105 cfu/mL. (ii) Reconventionalization with bacterial mixtures. For the preparation of Mix10, Mix9, Mix8 and all other mixes used, equimolar mixtures were prepared by
adding each bacterial species at initial concentration to 5.107 cfu/mL. Each bacterial mixture suspension was added to culture flasks containing germ-free fish at a final concentration of
5.105 cfu/mL.
F. columnare strains (Supplementary Table S1) were grown overnight in TYES broth at 28 °C. Then, 2 mL of culture were pelleted (10,000 rpm for 5 min) and washed once in sterile water. GF
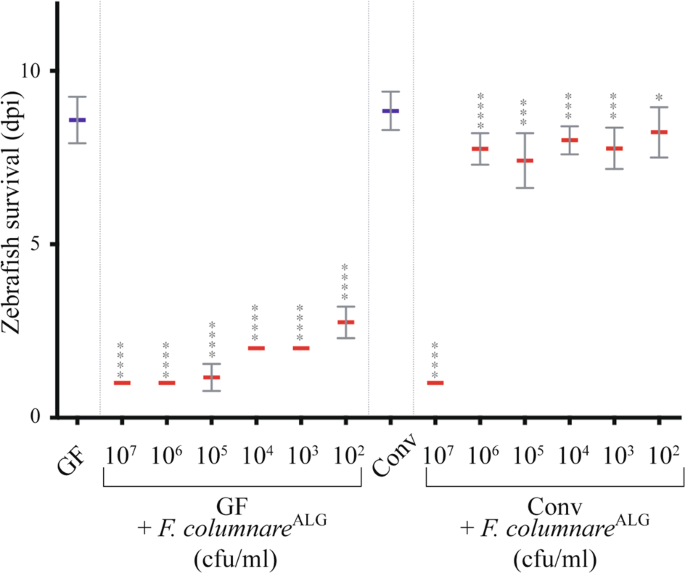
zebrafish were brought in contact with the tested pathogens at 6 dpf for 3 h by immersion in culture flasks with bacterial doses ranging from 5.102 to 5.107 cfu/mL. Fish were then
transferred to individual wells of 24-well plates, containing 2 mL of water and 50 μL of freshly prepared GF T. thermophila per well. Mortality was monitored daily as described in [22], and
measured in days post infection (dpi), with 0 dpi corresponding to the infection day, i.e. 6 dpf-old larvae. As few as 54 ± 9 cfu/larva of F. columnare were recovered from infected larvae.
All zebrafish experiments were stopped at day 9 post-infection and zebrafish were euthanized with tricaine (MS-222) (Sigma-Aldrich #E10521). Each experiment was repeated at least 3 times and
between 10 and 15 larvae were used per condition and per experiment.
Conventional zebrafish eggs were collected in 50 mL Falcon tubes from the following facilities: Facility 1 - zebrafish facility in Hospital Robert Debré, Paris; Facility 2 - Jussieu
zebrafish facility A2, University Paris 6; Facility 3 - Jussieu zebrafish facility C8 (UMR7622), University Paris 6; Facility 4- AMAGEN commercial facility, Gif sur Yvette; Larvae were
treated with the same rearing conditions, sterilization and infection procedures used in the Institut Pasteur facility.
Zebrafish were euthanized with tricaine (MS-222) (Sigma-Aldrich #E10521) at 0.3 mg/mL for 10 min. Then they were washed in 3 different baths of sterile PBS-0.1% Tween to remove bacteria
loosely attached to the skin. Finally, they were transferred to tubes containing calibrated glass beads (acid-washed, 425 μm to 600 μm, SIGMA-ALDRICH #G8772) and 500 μL of autoclaved PBS.
They were homogenized using FastPrep Cell Disrupter (BIO101/FP120 QBioGene) for 45 s at maximum speed (6.5 m/s). Finally, serial dilutions of recovered suspension were spotted on TYES agar
and cfu were counted after 48 h of incubation at 28 °C.
Over 3 months, the experiment was run independently 3 times and 3 different batches of eggs were collected from different fish couples in different tanks. Larvae were reared as described
above. GF and Conv larvae were collected at 6 dpf and 11 dpf for each batch. Infected Conv larvae were exposed to F. columnareALG for 3 h by immersion as described above. For each
experimental group, triplicate pools of 10 larvae (one in each experimental batch) were euthanized, washed and lysed as above. Lysates were split into 3 aliquots, one for culture followed by
16S rRNA gene sequencing (A), one for 16S rRNA gene clone library generation and Sanger sequencing (B), and one for Illumina metabarcoding-based sequencing (C).
Lysates were serially diluted and immediately plated on R2A, TYES, LB, MacConkey, BHI, BCYE, TCBS and TSB agars and incubated at 28 oC for 24-72 h. For each agar, colony morphotypes were
documented, and colonies were picked and restreaked on the same agar in duplicate. In order to identify the individual morphotypes, individual colonies were picked for each identified
morphotype from each agar, vortexed in 200 μL DNA-free water and boiled for 20 min at 90 oC. Five μL of this bacterial suspension were used as template for colony PCR to amplify the 16S rRNA
gene with the universal primer pair for the Domain bacteria 8 f (5’-AGA GTT TGA TCC TGG CTC AG-3’) and 1492r (5’-GGT TAC CTT GTT ACG ACT T-3’). Each primer was used at a final concentration
of 0.2 μM in 50 μL reactions. PCR cycling conditions were - initial denaturation at 94 °C for 2 min, followed by 32 cycles of denaturation at 94 °C for 1 min, annealing at 56 °C for 1 min,
and extension at 72 °C for 2 min, with a final extension step at 72 °C for 10 min. 16S rRNA gene PCR products were verified on 1% agarose gels, purified with the QIAquick® PCR purification
kit and two PCR products for each morphotype were sent for sequencing (Eurofins, Ebersberg, Germany). 16S rRNA sequences were manually proofread, and sequences of low quality were removed
from the analysis. Primer sequences were trimmed, and sequences were compared to GenBank (NCBI) with BLAST, and to the Ribosomal Database Project with SeqMatch. For genus determination a 95%
similarity cut-off was used, for Operational Taxonomic Unit determination, a 98% cut-off was used.
Total DNA was extracted from the lysates with the Mobio PowerLyzer® Ultraclean® kit according to manufacturer’s instructions. Germ-free larvae and DNA-free water were also extracted as
control samples. Extracted genomic DNA was verified by Tris-acetate-EDTA-agarose gel electrophoresis (1%) stained with GelRed and quantified by applying 2.5 μL directly to a NanoDrop®
ND-1000 Spectrophotometer. The 16S rRNA gene was amplified by PCR with the primers 8 f and 1492r, and products checked and purified as described above. Here, we added 25–50 ng of DNA as
template to 50 μL reactions. Clone libraries were generated with the pGEM®-T Easy Vector system (Promega) according to manufacturer’s instructions. Presence of the cloned insert was
confirmed by colony PCR with vector primers gemsp6 (5’-GCT GCG ACT TCA CTA GTG AT-3’) and gemt7 (5’-GTG GCA GCG GGA ATT CGA T-3’). Clones with an insert of the correct size were purified as
above and sent for sequencing (Eurofins, Ebersberg, Germany). Blanks using DNA-free water as template were run for all procedures as controls. For the three independent runs of the
experiment, 10 Conv fish per condition (6 and 11 dpf, exposed or not to F. columnare) and per repeat were pooled. Each pool of 10 fish was sequenced separately, generating 3 replicates for
each condition (n = 12), resulting in a total of 857 clones. Clone library coverage was calculated with the following formula [1-(n1/N2)] x 100, where n1 is the number of singletons detected
in the clone library, and N2 is the total number of clones generated for this sample. Clone libraries were generated to a minimum coverage of 95%. Sequence analysis and identification was
carried out as above.
To identify the 16S rRNA gene diversity in our facility and fish collected from 4 other zebrafish facilities, fish were reared as described above. GF fish were sterilized as above, and
uninfected germ-free and conventional fish were collected at 6 dpf and 11 dpf. Infection was carried out as above with F. columnareALG for 3 h by bath immersion, followed by transfer to
clean water. Infected conventional fish were collected at 6 dpf 6 h after infection with F. columnare and at 11 dpf, the same as uninfected fish. GF infected larvae were only collected at 6
dpf 6 h post infection, as at 11 dpf all larvae had succumbed to infection. Triplicate pools of 10 larvae were euthanized, washed and lysed as above. Total DNA was extracted with the Mobio
PowerLyzer® Ultraclean® kit as described above and quantified with a NanoDrop® ND-1000 Spectrophotometer and sent to IMGM Laboratories GmbH (Germany) for Illumina sequencing. Primers
Bakt_341F (5’-CCTACGGGNGGCWGCAG-3’) and Bakt_805R (5’-GACTACHVGGGTATCTAATCC-3’), amplifying variable regions 3 and 4 of the 16S gene were used for amplification [43]. Each amplicon was
purified with solid phase reversible immobilization (SPRI) paramagnetic bead-based technology (AMPure XP beads, Beckman Coulter) with a Bead:DNA ratio of 0.7:1 (v/v) following manufacturers
instructions. Amplicons were normalized with the Sequal-Prep Kit (Life Technologies), so each sample contained approximately 1 ng/μl DNA. Samples, positive and negative controls were
generated in one library. The High Sensitivity DNA LabChip Kit was used on the 2100 Bioanalyzer system (both Agilent Technologies) to check the quality of the purified amplicon library. For
cluster generation and sequencing, MiSeq® reagents kit 500 cycles Nano v2 (Illumina Inc.) was used. Before sequencing, cluster generation by two-dimensional bridge amplification was
performed, followed by bidirectional sequencing, producing 2 × 250 bp paired-end (PE) reads.
MiSeq® Reporter 2.5.1.3 software was used for primary data analysis (signal processing, demultiplexing, trimming of adapter sequences). CLC Genomics Workbench 8.5.1 (Qiagen) was used for
read-merging, quality trimming, and QC reports and OTU definition were carried out in the CLC plugin Microbial Genomics module.
Larvae reconventionalized with Mix10 and infected with F. columnareALG at 6 dpf for 3 h were euthanized and washed. DNA was extracted from pools of 10 whole larvae or of pools of 10
intestinal tubes dissected with sterile surgical tweezer and subjected to Illumina 16S rRNA gene sequencing. GF larvae and dissected GF intestines were sampled as controls. As dissection of
the larval guts involved high animal loss and was a potential important contamination source, we proceeded with using entire larvae for the rest of the study.
Chromosomal DNA of the ten species composing the core of zebrafish larvae microbiota was extracted using the DNeasy Blood & Tissue kit (QIAGEN) including RNase treatment. DNA quality and
quantity were assessed on a NanoDrop ND-1000 spectrophotometer (Thermo Scientific).
DNA sequencing libraries were made using the Nextera DNA Library Preparation Kit (Illumina Inc.) and library quality was checked using the High Sensitivity DNA LabChip Kit on the Bioanalyzer
2100 (Agilent Technologies). Sequencing clusters were generated using the MiSeq reagents kit v2 500 cycles (Illumina Inc.) according to manufacturer’s instructions. DNA was sequenced at the
Helmholtz Centre for Infection Research by bidirectional sequencing, producing 2 × 250 bp paired-end reads. Between 1,108,578 and 2,914,480 reads per sample were retrieved with a median of
1,528,402. Reads were quality filtered, trimmed and adapters removed with trimmomatic 0.39 [44] and genomes assembled using SPAdes 3.14 [45].
Whole genome-based bacterial species identification was performed by the TrueBac ID system (v1.92, DB:20190603) [46]. Species-level identification was performed based on the algorithmic
cut-off set at 95% ANI when possible or when the 16S rRNA gene sequence similarity was >99 %.
Three independent experiments were run over 6 weeks with eggs collected from different fish couples from different tanks to monitor establishment and recovery. Larvae were reared, sterilized
and infected as above with the only difference that 75 cm3 culture flasks with vented caps (filled with 50 mL of sterile Volvic) were used to accommodate the larger number of larvae
required, as in each experiment. Larvae for time course Illumina sequencing were removed sequentially from the experiment that monitored the survival of the larvae. Animals were pooled (10
larvae for each time point/condition), euthanized, washed and lysed as described above and stored at −20o C until the end of the survival monitoring, and until all triplicates had been
collected.
In order to follow the establishment of the 10 core strains in the larvae, GF larvae were reconventionalized with an equiratio Mix10 as above. Re-convMix10 larvae were sampled at 4 dpf
immediately after addition of the 10 core species and then 20 min, 2 h, 4 h and 8 h after. Germ-free, conventional larvae and the inoculum were also sampled as controls.
Different doses of kanamycin (dose 1 = 200 µg/mL; dose 2 = 50 µg/mL; dose 3 = 25 µg/mL) and a penicillin/streptomycin antibiotic mix dose 1 = 250 µg/mL; dose 2 = 15.6 µg/mL were tested on
re-convMix10 4 dpf zebrafish larvae by adding them to the flask water to identify antibiotic treatments that were non-toxic to larvae but that caused dysbiosis.
After 16 h of treatment, antibiotics were extensively washed off with sterile water and larvae were challenged with F. columnareALG, leading to the death of all larvae – e.g. successful
abolition of colonization resistance with best results in all repeats with 250 µg/mL penicillin/streptomycin and 50 µg/mL kanamycin as antibiotic treatment.
As above, after 8 h of incubation, 4 dpf re-convMix10 larvae were treated with 250 µg/mL penicillin/streptomycin and 50 µg/mL kanamycin for 16 h. Antibiotics were extensively washed off and
larvae were now left to recover in sterile water for 24 h to assess resilience of the bacterial community. Samples (pools of 10 larvae) were taken at 3 h, 6 h, 12 h, 18 h, and 24 h during
recovery and sent for 16S rRNA Illumina sequencing. Larvae were then challenged at 6 dpf with F. columnareALG for 3 h and survival was monitored daily for 9 days post-infection. All time
course samples were sequenced by IMGM Laboratories GmbH, as described above.
16S RNA analysis was performed with SHAMAN [47]. Library adapters, primer sequences, and base pairs occurring at 5’ and 3’ends with a Phred quality score