Innate and adaptive signals enhance differentiation and expansion of dual-antibody autoreactive b cells in lupus
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Autoreactive B cells have a major function in autoimmunity. A small subset of B cells expressing two distinct B-cell-antigen-receptors (B2R cells) is elevated in many patients with
systematic lupus erythematosus (SLE) and in the MRL(/_lpr_) mouse model of lupus, and is often autoreactive. Here we show, using RNAseq and in vitro and in vivo analyses, signals that are
required for promoting B2R cell numbers and effector function in autoimmune mice. Compared with conventional B cells, B2R cells are more responsive to Toll-like receptor 7/9 and type I/II
interferon treatment, display higher levels of MHCII and co-receptors, and depend on IL-21 for their homeostasis; moreover they expand better upon T cell-dependent antigen stimulation, and
mount a more robust memory response, which are characteristics essential for enhanced (auto)immune responses. Our findings thus provide insights on the stimuli for the expansion of an
autoreactive B cell subset that may contribute to the etiology of SLE. SIMILAR CONTENT BEING VIEWED BY OTHERS TOLL-LIKE RECEPTOR SIGNALLING IN B CELLS DURING SYSTEMIC LUPUS ERYTHEMATOSUS
Article 18 December 2020 THE ESSENTIAL ROLES OF MEMORY B CELLS IN THE PATHOGENESIS OF SYSTEMIC LUPUS ERYTHEMATOSUS Article 07 November 2024 ANTIGEN PRESENTATION BY B CELLS ENABLES EPITOPE
SPREADING ACROSS AN MHC BARRIER Article Open access 31 October 2023 INTRODUCTION A hallmark of systemic lupus erythematosus (SLE) is a breach in B cell tolerance that results in the
production of autoantibodies. Autoantibody-producing B cells play additional pathogenic roles in the progression of SLE by secreting inflammatory cytokines and directly activating pathogenic
T cells1,2,3. Autoreactive B cells originate from the bone marrow via expression of newly rearranged germline immunoglobulin (Ig) genes but also arise from peripheral lymphoid tissue
subsequent to the somatic hypermutation of Ig genes. During the development of B cells in the bone marrow, random Ig gene rearrangements generate a large number of autoreactive B cells4,5.
However, at least half of these cells are immediately eliminated by receptor editing (i.e., secondary Ig gene recombination) or clonal deletion6,7,8. Receptor editing most often results in
the elimination of the autoreactive specificity displayed by the B cell antigen receptor (BCR) and the expression of a new nonautoreactive BCR. However, receptor editing can also lead to the
generation of dual-reactive (B2R) B cells: B cells co-expressing two different heavy (H) or light (L, κ or λ) Ig chains9 and, thus, two different BCRs. These BCRs are comprised of the
initial autoreactive antigen receptor and a new nonautoreactive receptor most often based on the association of the original H chain with a new L chain. B2R cells have a demonstrated ability
to bypass tolerance checkpoints and escape to the periphery where they are more or less regulated by mechanisms of peripheral tolerance, depending on the genetic background and the level of
autoreactivity of the B cell clone10,11,12,13,14,15,16. Elegant recent studies indicate that many of the B cells that participate in disease flares in lupus patients are naive B cells with
germline Igs17 and they are therefore generated from de novo B cell development, as opposed to arising from a germinal center (GC) reaction. Thus, the establishment of central (bone marrow)
B cell tolerance is of vital importance for the prevention and/or control of autoimmunity in humans. B2R cells are observed in both healthy mice and humans at a frequency of less than 3% of
all B cells14,18,19,20,21,22. Our previous findings have shown that the fate of B2R cells can be different in an autoimmune background; here these cells expand with disease and show high
level of activation23. To study B2R cells we have employed the use of congenic mice bearing a gene targeted human _Ig Cκ_ allele24 on a healthy (CB17) or an autoimmune (MRL) genetic
background23. These _Igk__m/h_ mice allow for the detection of B cells co-expressing two different κ chains (dual-κ) within a wild-type Ig repertoire. We have previously found that dual-κ B
cells accumulate in both MRL and MRL/_lpr_ mice with age, while this is not observed in the non-autoimmune CB17 mice23. Furthermore, dual-κ B cell enrichment in MRL(/_lpr_) mice correlates
with disease progression and with the appearance of autoantibodies. These B2R cells are particularly enriched in the effector plasmablast and IgG+ memory B cell compartments of MRL(/_lpr_)
mice, where they represent up to 50% of cells in older mice. Overall, dual-κ B cells are more autoreactive, express higher levels of activation markers, and secrete larger amounts of
autoantibodies than single-κ B cells23. Interestingly, dual-κ B cells do not display these characteristics in NZB/NZW lupus mice25 and, thus, their association with lupus-like disease is
likely dependent on genetic polymorphisms. Similar to what has been described in lupus mice, a recent study found that B2R cells co-expressing κ and λ, are expanded in a subset (about 40%)
of SLE patients26. This suggests B2R cells are a significant player of SLE pathogenesis and that, gaining a better understanding of their biology, is important. In this study we exploited
our dual-κ autoimmune mouse model (MRL/_lpr_-_Igk__m/h_) to identify molecular pathways that can drive the accumulation and activation of B2R cells in murine lupus. We show that a
significant difference exists between single and dual-κ B cells both functionally and at the transcriptome level. Overall, our data indicate that both T cell-mediated signals and innate
stimuli, together and independently, play a role in the enrichment, activation, and differentiation of dual-κ B cells during lupus-like disease in MRL/_lpr_ mice. RESULTS MRL/_LPR_ DUAL-Κ B
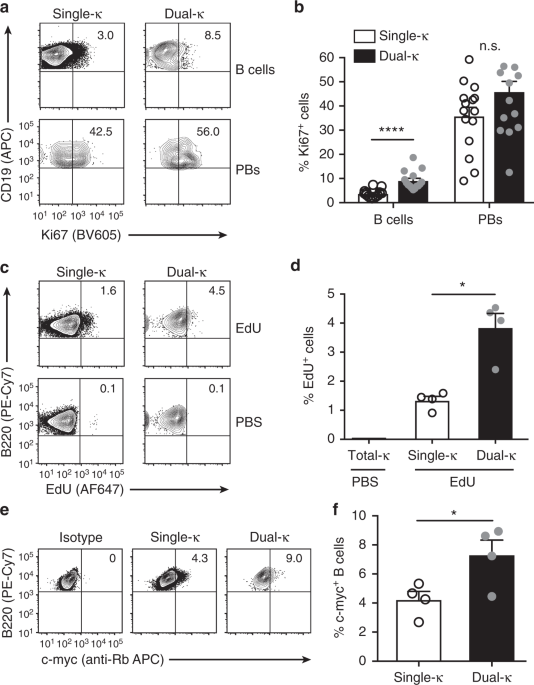
CELLS PROLIFERATE MORE THAN SINGLE-Κ CELLS We have previously shown that dual-κ B cells from MRL and MRL/_lpr_ lupus mice are enriched in the plasmablast cell compartment23, which is a stage
at which cells are actively proliferating. To evaluate whether this plasmablast enrichment is due to an increased proliferative ability of dual-κ relative to single-κ B cells, we analyzed
the expression of the proliferation marker Ki67 and the frequency of EdU incorporation, as readouts for active cell division. The frequency of Ki67+ dual-κ CD138– B cells was 2–3-fold
greater than that of single-κ cells in the spleen of naive MRL/_lpr_-_Igk__m/h_ mice, while it was similar in the CD138+ plasmablast compartment (Fig. 1a, b and Supplementary Fig. 1a, b).
Furthermore, 24 h after the injection of EdU, the frequency of EdU+ dual-κ splenic B cells was 3-fold higher than that of single-κ B cells (Fig. 1c, d and Supplementary Fig. 1a–c), thus
indicating enhanced active proliferation of dual-κ B cells in vivo. Previous studies have shown that c-Myc is rapidly upregulated by mature B cells following BCR engagement27 and that this
event leads to cell proliferation28. In line with the higher proliferation rates described above, a larger frequency of dual-κ B cells than single-κ B cells expressed c-Myc protein (Fig. 1e,
f and Supplementary Fig. 1a–d). Collectively, these results indicate that under homeostatic conditions, dual-κ B cells of MRL_/lpr_ mice proliferate at a higher rate than single-κ B cells
and possibly as a consequence of antigen engagement and c-Myc expression. RNASEQ AND PATHWAY ANALYSES OF SINGLE AND DUAL-Κ B CELLS To identify genes and pathways involved in the enhanced
activation and proliferation of dual-κ B cells in murine lupus, we performed RNAseq analyses of single and dual-κ B cells from MRL/_lpr-Igk__m/h_ mice. Because dual-κ B cells are biased
toward the marginal zone (MZ) subset23, in order to prevent an artificial skewing we analyzed single and dual-κ follicular (FO) and MZ splenic B cell populations, sorted as described in
Supplementary Fig. 2a. RNAseq analyses identified 1938 and 446 genes (false discovery rate, FDR ≤ 0.05) in FO and MZ B cells, respectively, that were differentially expressed in dual-κ B
cells relative to single-κ B cells (Fig. 2a). The overexpression of _Ig Vκ_ genes and not of _CD19_ and _CD79A_ genes in dual-κ cell samples, verified the accuracy of the cells used for the
RNAseq data (Fig. 2b). In addition, _Jκ1_ RNA amount was decreased in dual-κ B cells while _Jκ5_ was increased (Fig. 2c), a result that is consistent with higher frequency of receptor
editing29. We also analyzed the abundance of _CD138_ and _IgG_1, _IgG2_b, and _IgG_2c transcripts in the FO B cell samples to estimate the level of contamination by plasmablasts and
IgG-switched cells. One of the samples had a larger amount of these transcripts, but the contamination was generally similar in the dual-κ and single-κ samples (Supplementary Fig. 2b). We
next used ingenuity pathway analysis (IPA) to identify pathways that are specifically guiding the biology of dual-κ B cells. The differentially enriched pathways with particular
immunological significance include BAFF and APRIL-mediated signaling, Toll-like receptor signaling, IL-6 signaling, MAPK signaling, and antigen presentation, for both FO and MZ B cells (Fig.
2d). The top 30 pathways detected by IPA in FO B cells (-log(_p_-value) > 6.05) included pathways involved in antigen and cytokine receptor signaling, such as B Cell Receptor, TNFR1/2,
mTOR, PI3K/AKT, CD27, NFAT, JAK/Stat, and JAK1, JAK2, and TYK2 signaling in interferon. On the other hand, the pathways with B-cell relevance detected in MZ B cells among the top 30
(-log(_p_-value) > 4.53) were role of pattern recognition receptors in recognition of bacteria and viruses, complement system, IL-6 signaling, and toll-like receptor signaling. Thus,
while there was overlap in the RNAseq data from FO and MZ B cells, there were also some distinctions. Results from these gene expression analyses suggest dual-κ B cells from MRL/_lpr_ mice
possess a differential ability to respond to T cell-dependent and independent signals relative to single-κ B cells. DUAL-Κ CELLS SHOW HIGHER RESPONSES TO INNATE STIMULI The TLR and IFN I and
II pathways are of particular relevance for the onset and progression of lupus disease2,30,31. Based on the RNAseq analysis, dual-κ FO B cells displayed differential expression of 21 out of
76 genes and 14 out of 36 genes in the TLR signaling and the interferon signaling pathways, respectively (Fig. 3a). To validate these results, we investigated in vitro responses of single
and dual-κ B cells to these innate stimuli. Untouched (CD43–) B cells isolated from the spleen of MRL/_lpr-Igk__m/h_ mice were cultured in the presence or absence of the TLR ligands CpG (for
TLR9), R848 (for TLR7), and LPS (for TLR4), or with IFN-α (type I) or IFN-γ (type II) to measure their contribution to the relative frequency and activation of single and dual-κ B cells.
TLR7 and TLR9 stimulation significantly expanded dual-κ B cells over single-κ B cells, while TLR4 stimulation (LPS) had no effect (Fig. 3b and Supplementary Fig. 3a, b). Despite this
difference, single and dual-κ B cells similarly increased CD69 expression (Fig. 3c), suggesting that TLR7 and TLR9 signaling preferentially promotes the expansion, but not the activation of
dual-κ B cells relative to single-κ B cells. In contrast, culturing B cells with either IFN-α or IFN-γ induced significantly higher expression of CD69 (Fig. 3d and Supplementary Fig. 3a) and
CD86 (Supplementary Fig. 3c) on dual-κ relative to single-κ B cells, although neither IFN altered the relative frequency of dual-κ B cells (Fig. 3e). This indicates that IFNs I and II may
preferentially promote the activation and not the expansion of dual-κ B cells. The pathway analysis also highlighted a differential activation of BAFF and APRIL signaling, but incubation
with these two B cell survival factors did not promote any preferential expansion of dual-κ B cells (Supplementary Fig. 3d, e). We have previously shown that the splenic dual-κ B cell
population of MRL/_lpr_ mice holds a larger frequency of activated B cells than the single-κ population23. We wondered if MRL/_lpr_ dual-κ B cells display an increased activation starting
from their origin, and whether their increased activation accounts for their differential responses to TLR agonists and IFNs described above. To address the first issue we analyzed the
relative expression of the CD80 and CD86 activation markers on immature B cells of mice at 8 and 14 weeks of age. The expression of CD80 and CD86 on newly generated dual-κ B cells was larger
than that on single-κ B cells, while there was little or no differential expression of CD138 (Supplementary Fig. 4a, b). This suggests that from their onset, dual-κ B cells are inherently
more activated. To directly test whether the increased activation of dual-κ B cells skewed the in vitro responses to innate stimuli, we analyzed the responses of untouched naive B cells,
cells that were magnetically sorted from the spleen as CD43–CD80–CD86– (Supplementary Fig. 4c). Even when comparing the naive B cell population, dual-κ B cells displayed enhanced responses
(increased frequency and/or activation) to CpG, R848, and IFN-α relative to single-κ B cells (Fig. 3f, g). Together, these data indicate that acute innate stimuli such as TLR and interferon
receptor signaling play a role in the preferential enrichment and/or the increased activation of dual-κ B cells in MRL/_lpr_ mice. DUAL-Κ B CELLS DISPLAY HIGHER AMOUNTS OF CO-RECEPTORS IPA
indicated an enrichment of the antigen presentation pathway in dual-κ FO B cells (Fig. 2d). To assess the role T cells may play in the activation of dual-κ B cells, we analyzed the
expression of several co-receptors that B cells use for cognate engagement with T cells. In agreement with the RNAseq analysis, dual-κ B cells expressed significantly higher amounts of
surface MHCII proteins (Fig. 4a, b). This difference was more pronounced in the FO than the MZ B cell compartment, whereas no difference was observed at the plasmablast stage (Fig. 4c). The
RNAseq analysis also reported increased _IL-21R_ RNA transcripts in dual-κ relative to single-κ FO B cells, but not in MZ B cells (Fig. 4d). This is of interest since IL-21 is a cytokine
that is secreted by T FO (and extra-FO) helper (TFH) cells to play a critical role in the formation of GC B cells, Ig class switched B cells, and plasma cells32,33. Indeed, dual-κ B cells
expressing IL-21R were at higher frequency (Fig. 4e, f) and expressed higher levels (Fig. 4g) of this receptor when compared to single-κ cells. Dual-κ B cells also showed significant
upregulation of the T cell co-stimulatory molecules CD80, CD86, and PD-L1 relative to single-κ cells (Fig. 4h, i and Supplementary Fig. 5a), while no difference was seen in the expression of
the B cell co-receptor CD19 (Fig. 4i). In contrast, expression of the co-stimulatory molecule CD40, which is crucial for receiving T cell help, was significantly lower when measured on
total dual-κ B cells (Fig. 4i), but this appeared to be due to an expansion of a CD40low cell subset (Fig. 4h) that we identified to consist of plasmablasts (Supplementary Fig. 5b). Indeed,
CD40 levels on CD40+ B cells were slightly significantly higher on dual-κ than single-κ cells (Fig. 4h, i). This difference appeared to be functional, since stimulation of B cells with
anti-CD40 antibodies resulted in higher levels of CD69 (but not CD86) on dual-κ B cells (Supplementary Fig. 5c). Cognate interaction of B and T cells leads to the formation of GCs.
Relatedly, the frequency of PNAhighCD38low GC B cells within the dual-κ population was more than double than that observed in the single-κ cell subset (Fig. 4j, k). IgD levels, which can be
downregulated on antigen-exposed B cells, where 10% decreased on FO dual-κ relative to single-κ cells, while IgM was slightly upregulated, but these differences were not significant
(Supplementary Fig. 5d, e). Overall, these data indicate that dual-κ B cells upregulate many if not most of the known adaptive co-stimulatory receptors and, thus, are poised to receive
increased T cell help. CONTRIBUTION OF IL-21R TO THE DIFFERENTIATION OF DUAL-Κ B CELLS The development of lupus disease is significantly inhibited in MRL/_lpr_ mice deficient for IL-21R34.
IL-21, a cytokine secreted by CD4 TFH cells, promotes the generation of plasma cells and the development of IgG class switched antibodies and autoantibodies32,33. Given the increased
expression of IL-21R by dual-κ B cells (Fig. 4e–g), we questioned whether IL-21 contributes to the positive selection of these cells into effector populations. To study this, we crossed
IL-21R deficient MRL/_lpr_ mice34 to MRL/_lpr-Igk__h/h_ to generate MRL/_lpr-Igk__m/h_-IL21R−/− (IL-21R KO) mice. In accordance with a previous report34, we observed a reduction of B cell
numbers in IL-21R-deficient MRL/_lpr-Igk__m/h_ mice. However, only the FO B cell numbers were decreased while MZ B cells were not affected (Supplementary Fig. 6a, b). In the absence of
IL-21R, the dual-κ FO and MZ B cells were still more activated than single-κ cells (Supplementary Fig. 6c), but their frequency was significantly decreased (Fig. 5a, b). This reduction was
already present at the immature/transitional B cell stage in the spleen (Fig. 5c), suggesting that the generation and/or initial expansion of immature dual-κ B cells depends on IL-21. In
agreement with previous studies34, the total numbers of CD138+CD44high plasmablasts were 2–3-fold reduced in IL-21R KO mice (Supplementary Fig. 7a, b), but this did not equally affect single
and dual-κ cells. In fact, there was a two-fold reduction in the relative frequency of dual-κ cells in the plasmablast compartment of IL-21R-deficient MRL/_lpr-Igk__m/h_ mice (Fig. 5d, e).
To investigate whether IL-21R signaling confers the enhanced proliferation observed in dual-κ B cells (Fig. 1d), we injected IL-21R sufficient and deficient animals with EdU and analyzed
cells 24 h later. Although dual-κ B cells displayed more proliferation than single-κ B cells even in the absence of IL-21R signaling, the difference was much smaller, and the extent of
proliferation was severely reduced, when compared to IL-21R sufficient animals (Fig. 5f). The contribution of IL-21 to the generation of IgG+ B cells is controversial, with reports
indicating reduced, similar, or enhanced IgG+ memory B cells in IL-21R KO mice32,33. We found that the total numbers of IgG+, κ+ memory B cells were similar in MRL/_lpr_ with or without
IL-21R ablation, though trended to higher numbers in KO mice (Supplementary Fig. 7c, d). Nevertheless, the frequency of dual-κ cells within the IgG+CD138– B cell compartment was reduced by
about 2–3-fold in IL-21R KO mice (Fig. 5g, h). Given that the FO compartment is a major precursor population for both plasmablasts and memory B cells, we questioned whether the reduced
frequency of dual-κ cells in the effector populations of IL-21R KO mice was due to the initial effect observed in the FO pool. To gauge this, we normalized the frequencies of dual-κ cells in
both effector populations to that of the respective FO B cell population in each mouse. Once normalized, we observed a significant reduction in the expansion of IL-21R KO dual-κ B cells
into the memory B cell pool, but not into the plasmablast compartment (Fig. 5i). This suggests that IL-21 plays an additional, or more prominent, role in the generation of dual-κ memory B
cells independent of its effect in establishing the FO B cell pool. Overall, these data indicate that IL-21 signaling significantly contributes to the entry and enrichment of dual-κ B cells
in the naive (FO and MZ), blasting, and memory B cell subsets of MRL/_lpr_ mice. ENHANCED T CELL-DEPENDENT RESPONSE OF DUAL-Κ CELLS Given dual-κ B cells overexpress many co-stimulatory
receptors used for cognate interaction with T cells, we next investigated whether these cells mount higher T cell-dependent antigen-specific responses. To address this, we compared the
response of single-κ and dual-κ B cells to phycoerythrin (PE), a well characterized T cell-dependent protein antigen model that allows relative straightforward identification of
antigen-reactive B cells by flow cytometry35. Because MRL/_lpr_ mice fail to respond to T cell-dependent antigens as disease progresses36, we immunized young (6–7-wk-old) MRL/_lpr-Igk__m/h_
mice with PE in Alum confirming that the PE-specific IgG antibody response is comparable in magnitude to that of non-autoimmune CB17-_Igk__m/h_ mice (Supplementary Fig. 8a). At day 7 after
PE/Alum immunization, the frequency of PE-binding single-κ and dual-κ B cells within each MRL/_lpr-Igk__m/h_ mouse was analyzed using flow cytometry as a measure of their response to antigen
(Fig. 6a and Supplementary Fig. 8b). To compare the responses in different experiments, we normalized the frequency of PE-responding single and dual-κ B cells in immunized mice to that of
single-κ B cells in PBS-injected mice within each experiment. On average, the frequency of PE-reactive single-κ B cells increased almost 10-fold following PE/Alum immunization (Fig. 6b). For
dual-κ B cells, this difference was >40-fold, suggesting an increased participation of this subset in the response to PE (Fig. 6b). However, we noticed that in naive (PBS-treated)
animals, the frequency of PE-binding B cells (the antigen-specific clonal precursor frequency) was already higher in the dual-κ than the single-κ cell subset (Fig. 6a, b). In fact, the ratio
of dual-κ to single-κ PE-reactive B cells did not change with PE/Alum immunization (Fig. 6c), indicating a similar ability of single and dual-κ B cells to expand during a primary T
cell-dependent antigen response. As shown above (Fig. 5d–f), IL-21-R signaling contributes to the proliferation of dual-κ B cells and their enrichment into the plasmablast subset in vivo.
Thus, we next asked whether the expansion of PE-reactive dual-κ B cells was dependent on IL-21. To address this, we immunized groups of IL-21R KO and WT MRL/_lpr-Igk__m/h_ mice with PE/Alum
and compared the expansion of PE-reactive dual-κ B cells to that of single-κ B cells in naive mice from related strains. In the absence of IL-21R, both single-κ and dual-κ B cells were able
to expand in response to PE (Fig. 6d), and this expansion was similar (4–6-fold) in all populations relative to their precursor frequency in naive mice, negating a role for IL-21 in this
process. Because dual-κ B cells display enhanced response to both TLR7 and TLR9 stimulation in vitro, we next investigated if TLR9 signaling increases the T cell-dependent response of dual-κ
B cells in vivo. Hence, MRL/_lpr-Igk__m/h_ mice were immunized with PE either in Alum or with the addition of CpG (Fig. 6a). As previously observed, there was a larger frequency of
PE-binding cells within the dual-κ cell population following PE/Alum immunization (Fig. 6e). However, in mice that received PE with CpG, there was an even greater expansion of PE-reactive
dual-κ B cells (Fig. 6e), which was doubled relative to single-κ cells (Fig. 6f). Finally, we next questioned whether the larger frequency of antigen-reactive dual-κ B cells that follows a
primary response to a T cell-dependent antigen leads to a superior memory response. Thus, we immunized 5 week-old MRL/_lpr-Igk__m/h_ mice with PE/Alum and, after 5 weeks, boosted these mice
with PE without adjuvant and analyzed PE-reactive B cells one week later (Fig. 6g). The response of memory B cells was demonstrated by a much higher frequency of PE-binding cells in mice
that received both primary and boost immunizations relative to those that received only primary or boost injections (Fig. 6h). In mice subjected to the memory protocol, the frequency of
dual-κ B cells binding PE was almost 2-fold higher than that of single-κ B cells (Fig. 6h), denoting a greater ability of dual-κ memory B cells to respond to a recall antigen. Together,
these data indicate that independent of IL-21, dual-κ B cells clonally expand to a higher degree in response to a T cell-dependent antigen relative to single-κ B cells. This increased clonal
expansion is partly due to a larger antigen-reactive clonal frequency in the naive population and partly to enhanced response to TLR agonists, when available. Furthermore, after a primary
antigen encounter, dual-κ B cells differentiate into memory B cells that display heightened response to a secondary antigen encounter. DISCUSSION SLE affects about 0.2% of the world’s
population and has no available cure37 and treatments are general immunosuppressants that have minimal specificity and unwarranted side effects. B cells represent a notable target for
disease control, but current B cell targeted therapies promote immunodeficiency and increase risk to infections because they do not discriminate between B cells promoting SLE vs. those
protecting from pathogens. Defining the characteristics of B cell subsets that specifically contribute to SLE pathogenesis, therefore, can lead to the development of therapies that
ameliorate autoimmunity while preserving immunological defense. We and others have previously shown that B cells that co-express two distinct IgL chains and thus two BCRs (B2R cells) are
significantly expanded in lupus-prone MRL and MRL/_lpr_ mice23 but not in NZB/NZW mice25. In the present work we indicate that T cell-dependent signals and innate stimuli, such as IL-21,
type I and II IFN, and TLR agonists, all play an important role in the activation, expansion, and effector function of B2R cells in the MRL/_lpr_ autoimmune background. In addition to
secreting antibodies, B cells function as antigen-presenting cells and, as such, can be involved in the activation of pathogenic T cells in diverse autoimmune diseases34,38,39,40,41. We show
that dual-κ B cells of MRL/_lpr_ mice express much higher levels of MHCII, suggesting they may be better able to present antigen to CD4 T cells. This is of interest in the context of murine
lupus, as recent work has shown decreased T cell activation and disease symptoms in MRL/_lpr_ mice bearing MHCII-deficient B cells42. Dual-κ B cells express greater amounts of additional
surface receptors important for cognate engagement with T cells. Among these co-receptors we were intrigued by IL-21R, as IL-21 is an important T cell-derived soluble factor that aids the
generation of GC B cells, Ig class switched plasma cells and, potentially, memory B cells32,33. Moreover, IL-21 blockade and ablation of IL-21 signaling in lupus-prone mice ameliorate
disease symptoms by reducing total B cells, plasmablasts and autoantibody titers34,43,44. We show in our study that deficiency in IL-21R signaling reduces the frequency of dual-κ B cells in
the naive (FO and MZ), the plasmablast and the IgG+ memory B cell subsets of MRL/_lpr_ mice, indicating dual-κ B cells have an enhanced dependency on IL-21 for their maturation. The
reduction of dual-κ B cells in different subsets may have different explanations. For instance, dual-κ B cells were already decreased in the newly generated (immature and transitional)
splenic B cell fractions of IL-21R KO mice, possibly because IL-21R is expressed during bone marrow B cell development and plays a role in the generation of immature/transitional B cells45.
Thus, the reduced frequency of dual-κ B cells in the naive mature B cell population of IL-21R KO mice may be simply the result of an increased dependency of dual-κ B cells on IL-21 during
their earlier development. In regard to plasmablasts, the two-fold reduction present in IL-21R KO mice was equivalent to that observed in the naive FO B cell compartment leading us to
suspect that these events may be connected. However, there could be alternative explanations. Based on reduced EdU incorporation, dual-κ B cells are less proliferative in IL-21R KO than
wild-type mice, possibly leading to reduced blasting and, thus, plasmablast formation. Furthermore, IL-21R signaling in B cells is known to promote the generation of plasma cells in synergy
with CD40 signaling32,33, both of which, we show, are amplified in dual-κ (relative to single-κ) FO B cells. These observations suggest that dual-κ cells may have a better ability to
differentiate into plasmablasts in response to IL-21 and CD40. The significant reduction of dual-κ memory B cells in IL-21R-deficient mice is of particular interest since we otherwise
observed an upward trend, though not significant, of total memory B cells in the absence of IL-21 signaling. Dual-κ B cells were decreased in the memory compartment of IL-21R KO mice to a
greater extent relative to the initial effect observed in FO B cells, leading us to conclude that the enrichment of dual-κ B cells into the memory cell subset is crucially dependent on
IL-21R signaling. Presently, we cannot exclude that any of the differences observed in the frequency of dual-κ B cells in IL-21R KO mice are B cell-extrinsic and/or due to the absence of
autoimmunity in these mice. Dual-κ B cells also display a two-to-three fold higher expression of CD80, CD86, and PD-L1, which are co-receptors that promote TFH help and GC B cell
survival46,47. Indeed, the frequencies of GC B cells, cMyc+ cells, and proliferating cells were higher within the dual-κ cell subset, suggesting more frequent antigen responses and T cell
help. Interestingly, this was not apparent during the initial B cell response to PE immunization. In fact, immunizations with the T cell-dependent antigen PE in Alum elicited the same
magnitude of clonal expansion by both single and dual-κ B cells, and regardless of IL-21R signaling. Despite a similar expansion, the response of dual-κ B cells to the PE protein antigen was
amplified relative to that of single-κ B cells, but this amplification was accounted for by an increased antigen-reactive clonal frequency in the naive dual-κ B cell population. This
difference might be due to the expression of two Ig H + L chain combinations in each dual-κ cell, which doubles the chances of reactivity with any antigen, or to the increased expression of
autoreactive and cross-reactive specificities, which has been reported21,23. It is likely that the system we employed to investigate B cell response to antigen, which is the frequency of
cells binding antigen at day 7, does not reflect many of the effects mediated by T cell help, such as increased generation and survival of GC B cells, plasmablasts, and memory B cells.
Indeed, when we compared single and dual-κ B cells during a memory response to PE, we observed a much larger expansion (i.e., response) of dual-κ cells after boosting. This strongly
indicates that during the primary response dual-κ B cells differentiate into memory B cells in larger numbers and possibly because of enhanced T cell help. The activation and differentiation
of autoreactive B cells can also occur independent of T cell help2. Toll-like receptor signaling, particularly endosomal TLR7/TLR9, is required for the production of anti-nuclear antibodies
and the amplification of (auto)Ab responses48,49,50. IPA together with in vitro studies indicated a differential ability of dual-κ B cells to respond to TLR7/TLR9 agonists. Indeed, we show
that immunization of PE with the TLR9 agonist CpG leads to a two-fold greater expansion of PE-reactive dual-κ B cells in vivo relative to that elicited by Alum or in single-κ B cells,
corroborating the increased expansion to TLR7&9 agonists observed in vitro. Why dual-κ B cells display enhanced activity of the TLR7&9 pathways is unclear, but we speculate this may
be related to the larger frequency of dual-κ cells binding nuclear antigens23. Indeed, the continual internalization of nuclear self-antigens would be expected to lead to an enhanced
stimulation and activity of TLR7&9 signaling pathways. The IPA also revealed an elevated activity of IFN I and II signaling pathways in dual-κ B cells, which was confirmed by finding a
larger frequency of activated dual-κ (than single-κ) cells in B cell cultures treated with IFN-α or IFN-γ. Gene expression studies have identified a type I IFN signature in SLE
patients51,52, and this cytokine exacerbates autoimmunity also in mice53, where it can sensitize B cells to TLR7&9 signaling54,55. Moreover, recent studies have demonstrated that B
cell-intrinsic IFN-γ receptor signaling promotes spontaneous GCs, autoantibodies, and systemic autoimmunity in two lupus-prone mouse strains54,56 and potentially in humans54. We propose that
these distinct factors, stronger TLR and IFN signaling pathways, together with a higher frequency of reactivity for nuclear antigens, play a major role in the enrichment of (autoreactive)
dual-κ B cells in the mature naive, GC, plasmablasts and memory B cell compartments in mice with lupus. Our RNAseq analyses were performed on B cells displaying a FO (CD21+CD21lowCD24low) or
a MZ (CD21highCD1dhigh) phenotype. It may be argued that the results of the FO cell analyses could have been partially affected by the presence of memory B cells and plasmablasts, which are
in larger numbers in the dual-κ cell subset, and could display a FO phenotype. However, our data does not support this argument. In fact, although the FO B cell samples were likely
contaminated by memory B cells and plasmablasts, the analysis of IgG and CD138 transcripts does not support a larger contamination in the dual-κ cell samples (Supplementary Fig. 2b).
Furthermore, when we cultured naive (CD43–CD80–CD86–) B cells with TLR7&9 agonists or IFN-α, the responses of dual-κ cells were still above those of single-κ cells. The transcriptome
analysis revealed that the expression of one vs. two different BCRs on the surface of MRL/_lpr_ B cells results in a differential gene expression of many molecules involved in BCR signaling,
especially in FO B cells. The three main MAPK signaling pathways (ERK, JNK, p38), which were differentially enriched in dual-κ over single-κ FO B cells, have been shown to be activated in B
cells of SLE patients57 and of mouse models of lupus58. IPA also identified a differential activation of the NF-κB signaling cascade, a pathway whose activation in B cells has been
associated with SLE58,59,60 and that spreads downstream of both TLR and CD40. Other signaling pathways that have been reported to be highly activated in B cells of lupus mice are the PI3K
and mTOR58, which were also enriched in dual-κ B cells. Activation of PI3K has been shown to break peripheral tolerance in anergic autoreactive B cells leading to their activation and
response61, and could similarly support the preferential expansion of autoreactive dual-κ B cell clones. We assume that the observed transcriptome differences in the activity of many
signaling pathways between single- and dual-κ B cells reflect, at least to a degree, increased self-antigen-mediated signaling by an autoreactive BCR. Interestingly, dual-κ B cells show a
higher degree of activation (CD80 and CD86 expression) already in immature and transitional B cell subsets. The markers we used to define immature/transitional B cells (lower CD23, IgD, and
CD21, and higher CD24) might also capture a small fraction of activated mature B cells. However, this fraction would be likely too small compared to immature/transitional B cells to account
for the observations we describe. Thus, we contend these findings indicate that dual-κ B cells distinguish from single-κ B cells starting at their origin, and that this difference magnifies
as disease and inflammation progress. Recently, increased frequency of allelically included (κ+λ+) B2R cells has also been described in about half of SLE patients in the UK26, elevating the
clinical significance of our findings in mice. Thus, increased frequency of B2R cells may be a marker of disease in a large subset of lupus patients, and this B cell subset may one day
represent a relevant cell target in lupus. While the dual-κ B cells present in MRL/_lpr_ mice serve as an excellent model to investigate the mechanistic aspects of B2R cell function in
lupus, this mouse model has its limitations as it does not take into account the genetic diversity that is present in the general human population. The MRL/_lpr_ mouse strain also carries an
inactivating mutation in Fas, which likely adds to the general B cell activation. Therefore, future studies will investigate if the molecular pathways and cell surface receptors
differentially expressed in B cells of MRL/_lpr_ mice are also present in B2R cells of autoimmune patients. Collectively, our results show that dual-κ B cells in the MRL/_lpr_ autoimmune
background, which are cells that are mostly autoreactive23, are also highly proliferative and activated, and they display a distinct gene signature that predisposes to a preferential
response to innate stimuli and T cell-derived signals leading to enhanced enrichment and differentiation into effector B cells. These unique characteristics are those expected in key B cell
players in autoimmune responses and explain the advantage dual-κ B cells display over single-κ B cells during lupus disease in MRL/_lpr_ mice. They also reveal potential targets for their
selective ablation in autoimmunity. METHODS MICE _Igk__m/h_ in the MRL/_lpr_ and CB17 genetic backgrounds have been previously described22,23. The CB17 background of CB17-_Igk__m/h_ mice is
actually a mixed CB17 x BL/6 × 129 genetic background. MRL/_lpr_-IL21R−/− mice34 were bred to MRL/_lpr-Igk__h/h_ to generate MRL/_lpr-Igk__m/h_-IL21R−/− mice. Both males and females
6–20-wk-of-age were used in this study. Animals were randomly assigned to groups based on availability, but considering their age and sex. Age was considered for severity of disease: disease
symptoms start around 10 weeks of age and increase with age. Disease in males is slightly delayed in MRL/_lpr_ mice and, thus, males assigned to groups were generally about two weeks older
than females in same groups. For immunization studies, younger mice were used because older MRL/_lpr_ mice do not respond to vaccination. The age of the animals used in analyses is reported
in figure legends. No animals were excluded from the analyses besides one MRL/_lpr_-IL21R−/− mouse that showed absolutely no response to PE immunization (i.e., frequency of PE+ B cells was
in the range of naive mice): based on our experience this was due to a failure of i.p. injection. Group sizes were chosen based upon knowledge of the variation of individual analyses and to
maximize the chances of uncovering statistically significant differences of the mean. All mice were housed in Specific Pathogen Free (SPF) conditions. All animal procedures were approved by
the NJH (AS2533) and UCD-AMC (B-105314(05)1E and B-105317(03)1E) Institutional Animal Care and Use Committees and carried out in accordance with approved guidelines. CELL PREPARATION
Single-cell suspension from spleen were prepared in RPMI (Gibco) supplemented with 3% FBS. To remove erythrocytes cells were incubated for 2 min with a buffer composed of 0.15 M NH4Cl4, 10
mM KHCO3, and 0.1 mM EDTA (pH 8.0). Cells were then centrifuged at 300× _g_ and washed with media. ANTIBODIES AND FLOW CYTOMETRIC ANALYSIS Single cells isolated from bone marrow and spleen
tissues were first incubated with a rat anti-mouse FcγRII/III antibody (2.4G2; homemade) in FACS buffer (PBS 1X with 3% FBS and 0.05% NaN3) for 15 min on ice to prevent subsequent unspecific
binding of staining antibodies on cells through their Fc portion. Cells (2–3 × 106 for each staining panel) were then incubated for 20 min on ice in 50 µl FACS buffer that included
antibodies against surface markers, starting with biotin-conjugated antibodies in a first step staining and then with a mix of fluorochrome-conjugated antibodies and streptavidin in a second
step staining. Cells were washed with FACS buffer before and after each step. Fluorescent monoclonal antibodies against B220 (clone RA3–6B2; fluorochromes PE-Cy7 and APC-Cy7 at dilution
1:200 or PerCP-Cy5.5 at 1:100), CD1d (1B1; PE at 1:500 or PerCP-Cy5.5 at 1:100), CD19 (1D3; BV510 or APC; 1:200), CD138 (281–2; PE; 1:200), CD21 (7E9; APC; 1:200), CD22 (OX-97; APC; 1:500),
CD23 (B3B4; BV786; 1:200), CD24 (M1/69; AF700 or APC-Cy7; 1:200), CD3e (145-2C11; PE at 1:300; PE-Cy7 at 1:100; APC-Cy7 at 1:100), CD38 (90; PE; 1:500), CD44 (IM7; PerCP-Cy5.5; 1:400), CD69
(H1.2F3; PE-Cy7; 1:200), CD80 (16-10A1; APC or BV605; 1:200), CD86 (GL1; APC or APC-Cy7; 1:200), IgD (11–26 c.2a; BUV395 or APC-Cy7; 1:200), IL-21R (4A9; PE; 1:200), and PD-L1 (10 F.9G2;
APC; 1:200) were purchased from BioLegend. Antibodies to MHCII I-Ak (11–5.2; PE; 1:400), IgG1 (A85-1; APC; 1:100), and IgG2a/2b (R2-40; conjugated in our lab to Dylight 650 ThermoFisher;
1:100) were purchased from BD. Goat polyclonal Fab fragment antibodies to mouse IgM (cat. #115-097-020; FITC; 1:200) were purchased from Jackson Labs. Antibodies to CD40 (clone 1C10; used at
1:200) were made and conjugated to Dylight 650 (ThermoFisher) in our lab. Staining for Igκ was performed using a fluorescent polyclonal goat Fab’ anti-human Igκ (Protos Immunoresearch cat.
#376; FITC; 1:400) or a biotin-conjugated rat Fab anti-mouse Igκ (187.1; used at 1:100) that was made in our lab26. Biotin-labeled antibodies were visualized with streptavidin
(eBioscience/ThermoFisher; cat. # S11222 PacBlue; 1:200). Peanut agglutinin (PNA;FITC; 1:1000) for the staining of GC B cells was purchased from Vector Laboratories. Intracellular staining
for Ki67 (BioLegend; clone 16A8; BV605; 1:50) was performed using the True-Nuclear Transcription Factor Buffer Set (BioLegend). To stain for c-Myc, cells were first fixed in 2%
paraformaldehyde and then permeabilized in 90% methanol. Cells were washed in PBS and then stained with rabbit anti-mouse c-Myc (D84C12, Cell Signaling; 1:100) followed by polyclonal donkey
anti-rabbit IgG (BioLegend; #406406; Dylight-649; 1:100). Total rabbit IgG (Southern Biotech; 1:2000) was used as an isotype control for c-Myc staining. To stain for PE-specific B cells, 20
× 106 splenocytes per sample were first incubated with 4 μg of PE (Prozyme) in 1 ml staining buffer (1% BSA, 2 mM EDTA) for 30 min on ice. Cells were then washed twice with FACS buffer and
surface staining was continued as described above. Dead cells were excluded from analyses either by 7-amino-actinomycin D (7AAD; eBioscience) or propidium iodide (Sigma-Aldrich)
incorporation, or based on forward and side scatter. Sample acquisition was done using an LSRII or LSRFortessa cytometers (BD) and analyzed with FlowJo software v10.4.1 (Tree Star). Data
were analyzed on a live and lymphoid cell gate (based on forward and side scatter), followed by a doublet cell exclusion gate to eliminate cell aggregates and doublets. The lymphoid gate was
at times set to capture activated cells displaying higher FSC and SSC, as these cells are frequent in autoimmune mice. The CD3+B220+ T cell population that is characteristic of MRL/_lpr_
mice was excluded from the analyses of B cells by either gating out CD3+ events, or by gating B cells as CD19+ or CD22+, or as B220+ in combination with Igκ+, depending on the panel. SORTING
OF SINGLE-Κ AND DUAL-Κ B CELLS Splenocytes were pooled from _N_ = 2–4 mice for each biological replicate. These cells were first depleted of CD3 cells by complement-mediated lysis in the
following way. Splenocytes were incubated with anti-CD3 antibodies (145-2C11, homemade) at a concentration of 0.4 µg/106 cells for 20 min on ice. Cells were then washed and resuspended in
media containing reconstituted Low-Tox Guinea Pig Complement (Cedarlane) at a 1:20 dilution and incubated for 30 min at 37 °C. Afterwards, the cells were washed twice with media and then
incubated with a rat anti-mouse FcγRII/III antibody (2.4G2; homemade) in FACS buffer for 15 min on ice to prevent subsequent unspecific binding of staining antibodies. Cells were then
stained with fluorochrome-labeled antibodies. Stained cells were sorted as single or dual-κ CD3–CD21+CD1dlowCD24low FO B cells or CD3–CD21highCD1dhigh MZ B cells (as shown in Supplementary
Fig. 2a). Sorting purity was generally >95% for FO B cells and >70% for MZ B cells in three independent sorting experiments, and numbers of sorted cells varied from 5 × 104 to 8 × 106
depending on the population (5 × 104–16 × 104 for dual-κ cells). Cell sorting was performed using an ICyte Synergy cell sorter (Sony). RNASEQ AND GENE PATHWAY ANALYSES Three independently
sorted populations of single-κ and dual-κ FO or MZ B cells (sorted as described above under Sorting) were used for RNAseq analyses. Total RNA was extracted using the RNeasy Micro Kit
(Qiagen) and quality was determined by a BioAnalyzer (Agilent Technologies). The RNA Integrity Number (RIN) for all samples was greater than 8. The RNA was processed for next-generation
sequencing (NGS) library construction as developed in the NJH Genomics Core Service facility for analysis with a Life Technologies (Carlsbad, CA, USA) Ion Proton NGS platform. A modified
Clontech SMARTer® Ultra™ Low Input RNA Kit for Sequencing - v3 (Mountain View, CA, USA) and modified Kapa Biosystems KAPA Hyper Prep Kit (Wilmington, MA, USA) were used. Briefly, library
construction started with 1 ng of RNA, followed by SMARTer 1st strand cDNA synthesis (Takara), full length dscDNA amplification by LD-PCR, followed by purification and validation. Afterward,
the samples were sheared using a Covaris focused-ultrasonicator, concentrated by vacuum, and then processed through the Kapa protocol. The Kapa protocol consisted of end repair and
A-tailing, adapter ligation, library amplification, and a post-amplification cleanup step. Once validated, the libraries were sequenced as barcoded-pooled samples on a Ion Torrent Proton
Sequencer (ThermoFisher Scientific) using a P1 Ion Proton chip (vP1.1.17) and the Torrent Suite software v5.0.2. Reads greater than 30 bp were mapped to the mouse mm10 reference genome with
STAR v2.4.1d62. Uniquely mapping reads were counted for Ensembl Release 78 gene annotations using HTSeq v0.6.0 in intersection-nonempty mode63. Statistical significance for the differential
gene expression between single and dual-κ B cells was evaluated using DESeq2 package v1.4.564 for the R statistical software v3.2.0 (R Core Team, 2014, Vienna, Austria) using the Wald test.
_P_-value adjustment was performed with the Benjamini and Hochberg false discovery rate method within DESeq265. We used a statistical model with variables for the tissue of origin (FO or
MZ), the receptor type (single- or dual-κ), and their interaction, as well as a variable for the three independently sorted splenocyte cell pools, to account for any batch effects. A FDR ≤
0.05 was used to evaluate differential expression of individual genes. Differentially expressed genes were evaluated based on the contrast FO or MZ single-κ vs. dual-κ cells. Pathway
analysis was performed with IPA, IPA (QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/). Genes with a FDR of ≤ 0.1 were selected as differentially
expressed for IPA analysis. Heat maps were generated in Morpheus [https://software.broadinstitute.org/morpheus/] using min/max row-scaled rlog values, where the expression values are mapped
to colors using the minimum and maximum of each row/gene independently. IN VITRO CELL CULTURE Spleen (untouched) B cells were enriched by negative selection with anti-CD43 beads (Miltenyi
Biotech) on an AutoMACS Pro Separator following manufacturer instructions. Enriched B cells (92–99% pure) were cultured in complete RPMI at 2 × 106 cells/ml for 24 or 48 h and then analyzed
by flow cytometry. Where appropriate, the cells were stimulated with 50 nM CpG ODN 1826 (IDT), 10 μg/ml LPS (Sigma-Aldrich), 1 μg/ml R848 (Invivogen), 100 U/ml recombinant mouse IFN-α11 (PBL
Assay Science), 50 ng/ml IFNγ (R&D Systems), 50 ng/ml BAFF (R&D Systems), 50 ng/ml APRIL (PeproTech), or 15 μg/ml anti-CD40 antibodies (IC10, made in house). To isolate naive B
cells for in vitro studies, splenocytes were stained with PE-labeled antibodies against CD80 (BD clone B7-1; 1:400) and CD86 (BD clone B7-2; 1:400) and then cells were negatively selected
with a combination of anti-PE and anti-CD43 beads (Miltenyi Biotech). Purity of enriched naive B cells was about 99%. To control for non-specific dual-κ B cell detection, which was an issue
with TLR stimulation, for each experimental condition we also cultured a 1:1 mixture of homozygous Ig_k__h/h_ and wild-type Ig_k__m/m_ (h + m) cells (Supplementary Fig. 3b) and these
background events were subtracted from those measured in TLR-stimulated h/m cultures. IMMUNIZATIONS AND ELISA For PE immunizations, mice were injected i.p. with 150 μg PE (Prozyme) mixed
with either equal volume of Alu-Gel-S (Aluminum hydroxide, SERVA) or 50 μg CpG ODN 1826 (Invivogen) in PBS, and euthanized 7 d later for analysis. For memory studies, the mice were boosted 5
weeks after the initial immunization, with 150 μg of PE in the absence of adjuvant, and euthanized 7 d after the boost. To measure anti-PE IgG serum antibodies, 96-well Nunc Immuno MaxiSorp
plates (Thermo Fisher Scientific) were coated with 5 μg/ml PE in PBS overnight at 4 °C. Plates were washed three times with PBS/0.5% Tween-20, blocked for 2 h at 37 °C with blocking buffer
(1% BSA in PBS), then washed again. Serum samples were diluted starting at 1:50 in blocking buffer, then three-fold serial dilutions were made in the same buffer and plates were incubated
for 2 h at 37 °C. Plates were washed, incubated with alkaline phosphatase-conjugated goat anti-mouse IgG antibodies (Southern Biotech; cat #1030-04) diluted 1:1000 in blocking buffer, for 1
h at 37 °C, then washed again. To develop plates, 1 mg/ml of alkaline phosphatase substrate (Sigma-Aldrich) diluted in developing buffer (1 M diethanolamine, 8.4 mM MgCl2, and 0.02% NaN3, pH
9.8) was added to the plates. Absorbance values were read at 405 nm with a VersaMax ELISA Reader (Molecular Devices). IN VIVO EDU PROLIFERATION STUDIES Mice were injected i.p. with 1 mg EdU
(Sigma-Aldrich) in PBS and euthanized 24 h later. Spleen cells were harvested from EdU treated and untreated mice and stained for surface markers with antibodies listed above under
“Antibodies and flow cytometric analysis”. Cells were then fixed and stained for EdU using the Click-iT Plus EdU Alexa Fluor 647 Flow Cytometry Assay Kit (Thermo Fisher Scientific) following
manufacturer’s instructions. Flow cytometric analysis was then performed on an LSR Fortessa (BD). STATISTICS With the exception of RNAseq data, statistical analyses were performed with
GraphPad Prism software. Statistical significance for normally distributed data was determined by two-tailed _t_ tests, adding the Welch’s correction for groups with unequal variance, while
data that were not normally distributed were analyzed with the non-parametric Mann–Whitney test. Statistical significance for RNAseq data was assessed using the Wald test with the Benjamini
and Hochberg adjustment, and this is described in more details above under “RNAseq and gene pathway analyses”. Data are represented as means ± SEM. Significance levels are labeled as *_P_
< 0.05, **_P_ < 0.01, ***_P_ < 0.001, ****_P_ < 0.0001; n.s. not significant (_P_ > 0.05). All experimental replicates are biological, not technical. Investigators were not
blinded to samples. DATA AVAILABILITY The RNAseq data have been deposited in NCBI’s Gene Expression Omnibus66 under the accession number GSE109147. All other data supporting the findings of
these studies are available within the paper and the supplementary information files and/or from the corresponding author upon reasonable request. REFERENCES * Nashi, E., Wang, Y. &
Diamond, B. The role of B cells in lupus pathogenesis. _Int. J. Biochem. Cell. Biol._ 42, 543–550 (2010). Article CAS Google Scholar * Shlomchik, M. J. Activating systemic autoimmunity:
B’s, T’s, and tolls. _Curr. Opin. Immunol._ 21, 626–633 (2009). Article CAS Google Scholar * Bird, A. K., Meednu, N. & Anolik, J. H. New insights into B cell biology in systemic lupus
erythematosus and Sjogren’s syndrome. _Curr. Opin. Rheumatol._ 27, 461–467 (2015). Article CAS Google Scholar * Wardemann, H. et al. Predominant autoantibody production by early human B
cell precursors. _Science_ 301, 1374–1377 (2003). Article ADS CAS Google Scholar * Grandien, A., Fucs, R., Nobrega, A., Andersson, J. & Coutinho, A. Negative selection of
multireactive B cell clones in normal adult mice. _Eur. J. Immunol._ 24, 1345–1352 (1994). Article CAS Google Scholar * Shlomchik, M. J. Sites and stages of autoreactive B cell activation
and regulation. _Immunity_ 28, 18–28 (2008). Article CAS Google Scholar * Goodnow, C. C., Sprent, J., Fazekas de St Groth, B. & Vinuesa, C. G. Cellular and genetic mechanisms of self
tolerance and autoimmunity. _Nature_ 435, 590–597 (2005). Article ADS CAS Google Scholar * Pelanda, R. & Torres, R.M. Central B-cell tolerance: Where selection begins. _Cold Spring
Harb. Perspect. Biol_. 4, a007146 (2012). Article Google Scholar * Pelanda, R. Dual immunoglobulin light chain B cells: Trojan horses of autoimmunity? _Curr Opin Immunol_ 27, 53–59
(2014). Article Google Scholar * Liu, S. et al. Receptor editing can lead to allelic inclusion and development of B cells that retain antibodies reacting with high avidity autoantigens.
_J. Immunol._ 175, 5067–5076 (2005). Article CAS Google Scholar * Li, H., Jiang, Y., Prak, E. L., Radic, M. & Weigert, M. Editors and editing of anti-DNA receptors. _Immunity_ 15,
947–957 (2001). Article CAS Google Scholar * Andrews, S. F. et al. Global analysis of B cell selection using an immunoglobulin light chain-mediated model of autoreactivity. _J. Exp. Med._
210, 125–142 (2013). Article CAS Google Scholar * Kenny, J. J. et al. Autoreactive B cells escape clonal deletion by expressing multiple antigen receptors. _J. Immunol._ 164, 4111–4119
(2000). Article CAS Google Scholar * Li, Y., Li, H. & Weigert, M. Autoreactive B cells in the marginal zone that express dual receptors. _J. Exp. Med._ 195, 181–188 (2002). Article
CAS Google Scholar * Gerdes, T. & Wabl, M. Autoreactivity and allelic inclusion in a B cell nuclear transfer mouse. _Nat. Immunol._ 5, 1282–1287 (2004). Article CAS Google Scholar *
Kalinina, O. et al. Alternative mechanisms of receptor editing in autoreactive B cells. _Proc. Natl Acad. Sci. USA_ 108, 7125–7130 (2011). Article ADS CAS Google Scholar * Tipton, C. M.
et al. Diversity, cellular origin and autoreactivity of antibody-secreting cell population expansions in acute systemic lupus erythematosus. _Nat. Immunol._ 16, 755–765 (2015). Article CAS
Google Scholar * Pauza, M. E., Rehmann, J. A. & LeBien, T. W. Unusual patterns of immunoglobulin gene rearrangement and expression during human B cell ontogeny: human B cells can
simultaneously express cell surface kappa and lambda light chains. _J. Exp. Med._ 178, 139–149 (1993). Article CAS Google Scholar * Giachino, C., Padovan, E. & Lanzavecchia, A.
kappa+lambda+dual receptor B cells are present in the human peripheral repertoire. _J. Exp. Med._ 181, 1245–1250 (1995). Article CAS Google Scholar * DeKosky, B. J. et al. In-depth
determination and analysis of the human paired heavy- and light-chain antibody repertoire. _Nat. Med._ 21, 86–91 (2015). Article CAS Google Scholar * Casellas, R. et al. Igkappa allelic
inclusion is a consequence of receptor editing. _J. Exp. Med._ 204, 153–160 (2007). Article CAS Google Scholar * Velez, M. G. et al. Ig allotypic inclusion does not prevent B cell
development or response. _J. Immunol._ 179, 1049–1057 (2007). Article CAS Google Scholar * Fournier, E. M. et al. Dual-reactive B cells are autoreactive and highly enriched in the
plasmablast and memory B cell subsets of autoimmune mice. _J. Exp. Med._ 209, 1797–1812 (2012). Article CAS Google Scholar * Casellas, R. et al. Contribution of receptor editing to the
antibody repertoire. _Science_ 291, 1541–1544 (2001). Article ADS CAS Google Scholar * Makdasi, E. & Eilat, D. L chain allelic inclusion does not increase autoreactivity in
lupus-prone New Zealand Black/New Zealand White mice. _J. Immunol._ 190, 1472–1480 (2013). Article CAS Google Scholar * Fraser, L. D. et al. Immunoglobulin light chain allelic inclusion
in systemic lupus erythematosus. _Eur. J. Immunol._ 45, 2409–2419 (2015). Article CAS Google Scholar * Leider, N. & Melamed, D. Differential c-Myc responsiveness to B cell receptor
ligation in B cell-negative selection. _J. Immunol._ 171, 2446–2452 (2003). Article CAS Google Scholar * Murn, J. et al. A Myc-regulated transcriptional network controls B-cell fate in
response to BCR triggering. _BMC Genom._ 10, 323 (2009). Article Google Scholar * Nemazee, D. & Weigert, M. Revising B cell receptors. _J. Exp. Med._ 191, 1813–1817 (2000). Article
CAS Google Scholar * Banchereau, R. et al. Personalized immunomonitoring uncovers molecular networks that stratify lupus patients. _Cell_ 165, 551–565 (2016). Article CAS Google Scholar
* Rawlings, D. J., Metzler, G., Wray-Dutra, M. & Jackson, S. W. Altered B cell signalling in autoimmunity. _Nat. Rev. Immunol._ 17, 421–436 (2017). Article CAS Google Scholar *
Zotos, D. & Tarlinton, D. M. Determining germinal centre B cell fate. _Trends Immunol._ 33, 281–288 (2012). Article CAS Google Scholar * Moens, L. & Tangye, S. G.
Cytokine-mediated regulation of plasma cell generation: IL-21 takes center stage. _Front. Immunol._ 5, 65 (2014). Article Google Scholar * Rankin, A. L. et al. IL-21 receptor is required
for the systemic accumulation of activated B and T lymphocytes in MRL/MpJ-Fas(lpr/lpr)/J mice. _J. Immunol._ 188, 1656–1667 (2012). Article CAS Google Scholar * Pape, K. A., Taylor, J.
J., Maul, R. W., Gearhart, P. J. & Jenkins, M. K. Different B cell populations mediate early and late memory during an endogenous immune response. _Science_ 331, 1203–1207 (2011).
Article ADS CAS Google Scholar * Park, C. L. et al. Isotypic profiles and other fine characteristics of immune responses to exogenous thymus-dependent and -independent antigens by mice
with lupus syndromes. _J. Immunol._ 130, 2161–2167 (1983). CAS PubMed Google Scholar * Rees, F., Doherty, M., Grainge, M. J., Lanyon, P. & Zhang, W. The worldwide incidence and
prevalence of systemic lupus erythematosus: a systematic review of epidemiological studies. _Rheumatol. (Oxf.)_ 56, 1945–1961 (2017). Article Google Scholar * Falcone, M., Lee, J.,
Patstone, G., Yeung, B. & Sarvetnick, N. B lymphocytes are crucial antigen-presenting cells in the pathogenic autoimmune response to GAD65 antigen in nonobese diabetic mice. _J.
Immunol._ 161, 1163–1168 (1998). CAS PubMed Google Scholar * Noorchashm, H. et al. I-Ag7-mediated antigen presentation by B lymphocytes is critical in overcoming a checkpoint in T cell
tolerance to islet beta cells of nonobese diabetic mice. _J. Immunol._ 163, 743–750 (1999). CAS PubMed Google Scholar * Wong, F. S. et al. Investigation of the role of B-cells in type 1
diabetes in the NOD mouse. _Diabetes_ 53, 2581–2587 (2004). Article CAS Google Scholar * Parker Harp, C. R. et al. B cell antigen presentation is sufficient to drive neuroinflammation in
an animal model of multiple sclerosis. _J. Immunol._ 194, 5077–5084 (2015). Article CAS Google Scholar * Giles, J. R., Kashgarian, M., Koni, P. A. & Shlomchik, M. J. B cell-specific
MHC Class II deletion reveals multiple nonredundant roles for B cell antigen presentation in murine lupus. _J. Immunol._ 195, 2571–2579 (2015). Article CAS Google Scholar * Bubier, J. A.
et al. A critical role for IL-21 receptor signaling in the pathogenesis of systemic lupus erythematosus in BXSB-Yaa mice. _Proc. Natl Acad. Sci. USA_ 106, 1518–1523 (2009). Article ADS CAS
Google Scholar * Herber, D. et al. IL-21 has a pathogenic role in a lupus-prone mouse model and its blockade with IL-21R.Fc reduces disease progression. _J. Immunol._ 178, 3822–3830
(2007). Article CAS Google Scholar * Simard, N. et al. Analysis of the role of IL-21 in development of murine B cell progenitors in the bone marrow. _J. Immunol._ 186, 5244–5253 (2011).
Article CAS Google Scholar * Good-Jacobson, K. L., Song, E., Anderson, S., Sharpe, A. H. & Shlomchik, M. J. CD80 expression on B cells regulates murine T follicular helper
development, germinal center B cell survival, and plasma cell generation. _J. Immunol._ 188, 4217–4225 (2012). Article CAS Google Scholar * Good-Jacobson, K. L. et al. PD-1 regulates
germinal center B cell survival and the formation and affinity of long-lived plasma cells. _Nat. Immunol._ 11, 535–542 (2010). Article CAS Google Scholar * Christensen, S. R. et al.
Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. _Immunity_ 25, 417–428 (2006). Article CAS
Google Scholar * Sang, A. et al. Activation of rheumatoid factor-specific B cells is antigen dependent and occurs preferentially outside of germinal centers in the lupus-prone NZM2410 mouse
model. _J. Immunol._ 193, 1609–1621 (2014). Article CAS Google Scholar * Rookhuizen, D. C. & DeFranco, A. L. Toll-like receptor 9 signaling acts on multiple elements of the germinal
center to enhance antibody responses. _Proc. Natl Acad. Sci. USA_ 111, E3224–E3233 (2014). Article ADS CAS Google Scholar * Psarras, A., Emery, P. & Vital, E. M. Type I
interferon-mediated autoimmune diseases: pathogenesis, diagnosis and targeted therapy. _Rheumatol. (Oxf.)_ 56, 1662–1675 (2017). Google Scholar * Pascual, V., Farkas, L. & Banchereau,
J. Systemic lupus erythematosus: all roads lead to type I interferons. _Curr. Opin. Immunol._ 18, 676–682 (2006). Article CAS Google Scholar * Perry, D., Sang, A., Yin, Y., Zheng, Y. Y.
& Morel, L. Murine models of systemic lupus erythematosus. _J. Biomed. Biotechnol._ 2011, 271694 (2011). Article Google Scholar * Jackson, S. W. et al. B cell IFN-gamma receptor
signaling promotes autoimmune germinal centers via cell-intrinsic induction of BCL-6. _J. Exp. Med._ 213, 733–750 (2016). Article CAS Google Scholar * Thibault, D. L. et al. Type I
interferon receptor controls B-cell expression of nucleic acid-sensing Toll-like receptors and autoantibody production in a murine model of lupus. _Arthritis Res. Ther._ 11, R112 (2009).
Article Google Scholar * Domeier, P. P. et al. IFN-gamma receptor and STAT1 signaling in B cells are central to spontaneous germinal center formation and autoimmunity. _J. Exp. Med._ 213,
715–732 (2016). Article CAS Google Scholar * Grammer, A. C., Fischer, R., Lee, O., Zhang, X. & Lipsky, P. E. Flow cytometric assessment of the signaling status of human B lymphocytes
from normal and autoimmune individuals. _Arthritis Res. Ther._ 6, 28–38 (2004). Article CAS Google Scholar * Wu, T. et al. Shared signaling networks active in B cells isolated from
genetically distinct mouse models of lupus. _J. Clin. Invest._ 117, 2186–2196 (2007). Article CAS Google Scholar * Pacheco, G. V. et al. Expression of TLR-7, MyD88, NF-kB, and INF-alpha
in B lymphocytes of Mayan women with systemic lupus erythematosus in Mexico. _Front. Immunol._ 7, 22 (2016). Article Google Scholar * Zhang, W. et al. Aberrant CD40-induced NF-kappaB
activation in human lupus B lymphocytes. _PLoS ONE_ 7, e41644 (2012). Article ADS CAS Google Scholar * Getahun, A., Beavers, N. A., Larson, S. R., Shlomchik, M. J. & Cambier, J. C.
Continuous inhibitory signaling by both SHP-1 and SHIP-1 pathways is required to maintain unresponsiveness of anergic B cells. _J. Exp. Med._ 213, 751–769 (2016). Article CAS Google
Scholar * Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. _Bioinformatics_ 29, 15–21 (2013). Article CAS Google Scholar * Anders, S., Pyl, P. T. & Huber, W. HTSeq—a
Python framework to work with high-throughput sequencing data. _Bioinformatics_ 31, 166–169 (2015). Article CAS Google Scholar * Love, M. I., Huber, W. & Anders, S. Moderated
estimation of fold change and dispersion for RNA-seq data with DESeq2. _Genome Biol._ 15, 550 (2014). Article Google Scholar * Benjamini, Y. & Hochberg, Y. Controlling the false
discovery rate: a practical and powerful approach to multiple testing . _J. Royal Stat. Soc. Ser. B (Methodol.)_ 57, 289–300 (1995). MathSciNet MATH Google Scholar * Edgar, R., Domrachev,
M. & Lash, A. E. Gene expression omnibus: NCBI gene expression and hybridization array data repository. _Nucleic Acids Res._ 30, 207–210 (2002). Article CAS Google Scholar Download
references ACKNOWLEDGEMENTS The authors acknowledge Pfizer as the source of the MRL/_lpr_-IL21R KO mice. We thank the NJH Genomics Core Service facility, and particularly Kendra Walton, for
their technical help with the RNASeq studies. We acknowledge the NJH and University of Colorado Department of Immunology and Microbiology flow cytometry facilities for cell sorting and flow
cytometry acquisition, and the Biological Resource Center at NJH and the Vivarium at the University of Colorado AMC for assistance with mouse husbandry. We are grateful to members of the
Pelanda and Torres laboratories for their advice and valuable discussions and to Dr. Kathy Pape (University of Minnesota) for her advice on PE immunization and PE-reactive B cell staining.
This study was supported by the National Institutes of Health grants AI052310 (to R.P.), AI052310-S1 (to R.P. and A.S.), AI052157 and AI078468 (to R.M.T.). It was also supported in part by a
grant to R.P. from the Lupus Research Institute, Inc., and by the Cancer Center Support Grant P30CA046934 for shared resource. A.S. was also partly supported by the T32 AI074491 training
grant. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Immunology and Microbiology, University of Colorado School of Medicine, Aurora, CO, 80045, USA Allison Sang, Jacob N.
Peterson, Brian P. O’Connor, Raul M. Torres & Roberta Pelanda * Center for Genes, Environment and Health, National Jewish Health, Denver, CO, 80206, USA Thomas Danhorn, Brian P. O’Connor
& Sonia M. Leach * Inflammation and Immunology, Pfizer Research, Cambridge, MA, 02140, USA Andrew L. Rankin * Immuno-Oncology Discovery, FivePrime Therapeutics, South San Francisco, CA,
94080, USA Andrew L. Rankin * Department of Biomedical Research, National Jewish Health, Denver, CO, 80206, USA Brian P. O’Connor, Sonia M. Leach, Raul M. Torres & Roberta Pelanda *
Department of Pediatrics, National Jewish Health, Denver, CO, 80206, USA Brian P. O’Connor Authors * Allison Sang View author publications You can also search for this author inPubMed Google
Scholar * Thomas Danhorn View author publications You can also search for this author inPubMed Google Scholar * Jacob N. Peterson View author publications You can also search for this
author inPubMed Google Scholar * Andrew L. Rankin View author publications You can also search for this author inPubMed Google Scholar * Brian P. O’Connor View author publications You can
also search for this author inPubMed Google Scholar * Sonia M. Leach View author publications You can also search for this author inPubMed Google Scholar * Raul M. Torres View author
publications You can also search for this author inPubMed Google Scholar * Roberta Pelanda View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS
R.P. and A.S. designed the experiments and interpreted the data. R.M.T. contributed to data interpretation and experimental design. A.S. performed the experiments with technical help from
J.N.P. B.P.O’.C. supervised the RNAseq data acquisition and S.M.L. and T.D. analyzed the RNAseq data. A.L.R. contributed the IL-21R KO MRL/_lpr_ mouse. A.S. and R.P. wrote the manuscript
with the editorial assistance of other authors. CORRESPONDING AUTHOR Correspondence to Roberta Pelanda. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests.
ADDITIONAL INFORMATION PUBLISHER'S NOTE: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. ELECTRONIC SUPPLEMENTARY
MATERIAL SUPPLEMENTARY INFORMATION PEER REVIEW FILE RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits
use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the
Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated
otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds
the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and
permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Sang, A., Danhorn, T., Peterson, J.N. _et al._ Innate and adaptive signals enhance differentiation and expansion of dual-antibody
autoreactive B cells in lupus. _Nat Commun_ 9, 3973 (2018). https://doi.org/10.1038/s41467-018-06293-z Download citation * Received: 25 January 2018 * Accepted: 10 August 2018 * Published:
28 September 2018 * DOI: https://doi.org/10.1038/s41467-018-06293-z SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a
shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative