Anti-tumour immunity induces aberrant peptide presentation in melanoma
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Extensive tumour inflammation, which is reflected by high levels of infiltrating T cells and interferon-γ (IFNγ) signalling, improves the response of patients with melanoma to
checkpoint immunotherapy1,2. Many tumours, however, escape by activating cellular pathways that lead to immunosuppression. One such mechanism is the production of tryptophan metabolites
along the kynurenine pathway by the enzyme indoleamine 2,3-dioxygenase 1 (IDO1), which is induced by IFNγ3,4,5. However, clinical trials using inhibition of IDO1 in combination with blockade
of the PD1 pathway in patients with melanoma did not improve the efficacy of treatment compared to PD1 pathway blockade alone6,7, pointing to an incomplete understanding of the role of IDO1
and the consequent degradation of tryptophan in mRNA translation and cancer progression. Here we used ribosome profiling in melanoma cells to investigate the effects of prolonged IFNγ
treatment on mRNA translation. Notably, we observed accumulations of ribosomes downstream of tryptophan codons, along with their expected stalling at the tryptophan codon. This suggested
that ribosomes bypass tryptophan codons in the absence of tryptophan. A detailed examination of these tryptophan-associated accumulations of ribosomes—which we term ‘W-bumps’—showed that
they were characterized by ribosomal frameshifting events. Consistently, reporter assays combined with proteomic and immunopeptidomic analyses demonstrated the induction of ribosomal
frameshifting, and the generation and presentation of aberrant _trans-_frame peptides at the cell surface after treatment with IFNγ. Priming of naive T cells from healthy donors with
aberrant peptides induced peptide-specific T cells. Together, our results suggest that IDO1-mediated depletion of tryptophan, which is induced by IFNγ, has a role in the immune recognition
of melanoma cells by contributing to diversification of the peptidome landscape. Access through your institution Buy or subscribe This is a preview of subscription content, access via your
institution ACCESS OPTIONS Access through your institution Access Nature and 54 other Nature Portfolio journals Get Nature+, our best-value online-access subscription $32.99 / 30 days cancel
any time Learn more Subscribe to this journal Receive 51 print issues and online access $199.00 per year only $3.90 per issue Learn more Buy this article * Purchase on SpringerLink *
Instant access to full article PDF Buy now Prices may be subject to local taxes which are calculated during checkout ADDITIONAL ACCESS OPTIONS: * Log in * Learn about institutional
subscriptions * Read our FAQs * Contact customer support SIMILAR CONTENT BEING VIEWED BY OTHERS ADAR1 MASKS THE CANCER IMMUNOTHERAPEUTIC PROMISE OF ZBP1-DRIVEN NECROPTOSIS Article 25 May
2022 BLOCKADE OF THE AHR RESTRICTS A TREG-MACROPHAGE SUPPRESSIVE AXIS INDUCED BY L-KYNURENINE Article Open access 11 August 2020 PARP14 INHIBITION RESTORES PD-1 IMMUNE CHECKPOINT INHIBITOR
RESPONSE FOLLOWING IFNΓ-DRIVEN ACQUIRED RESISTANCE IN PRECLINICAL CANCER MODELS Article Open access 26 September 2023 DATA AVAILABILITY Data have been deposited in the Gene Expression
Omnibus (GEO) with accession code GSE142822. The genomic data relevant to the MD55A3 cells are found in BioProject with accession code PRJNA316754 (sample ID 1M1). Proteomics and peptidomics
data have been deposited in the PRIDE repository78 with accession code PXD020224. The codes used in the study are available on GitHub (https://github.com/apataskar/bump_finder_example2 and
https://github.com/apataskar/accessory_scripts_manuscript). REFERENCES * Ayers, M. et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. _J. Clin. Invest_. 127,
2930–2940 (2017). PubMed PubMed Central Google Scholar * Ji, R. R. et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. _Cancer Immunol. Immunother_. 61,
1019–1031 (2012). CAS PubMed Google Scholar * Timosenko, E. et al. Nutritional stress induced by tryptophan-degrading enzymes results in ATF4-dependent reprogramming of the amino acid
transporter profile in tumor cells. _Cancer Res_. 76, 6193–6204 (2016). CAS PubMed PubMed Central Google Scholar * Zhai, L. et al. Molecular pathways: targeting IDO1 and other tryptophan
dioxygenases for cancer immunotherapy. _Clin. Cancer Res_. 21, 5427–5433 (2015). CAS PubMed PubMed Central Google Scholar * Amobi, A., Qian, F., Lugade, A. A. & Odunsi, K.
Tryptophan catabolism and cancer immunotherapy targeting IDO mediated immune suppression. _Adv. Exp. Med. Biol_. 1036, 129–144 (2017). CAS PubMed Google Scholar * Labadie, B. W., Bao, R.
& Luke, J. J. Reimagining IDO pathway inhibition in cancer immunotherapy via downstream focus on the tryptophan-kynurenine-aryl hydrocarbon axis. _Clin. Cancer Res_. 25, 1462–1471
(2019). CAS PubMed Google Scholar * Günther, J., Däbritz, J. & Wirthgen, E. Limitations and off-target effects of tryptophan-related IDO inhibitors in cancer treatment. _Front.
Immunol_. 10, 1801 (2019). PubMed PubMed Central Google Scholar * Battu, S., Minhas, G., Mishra, A. & Khan, N. Amino acid sensing via general control nonderepressible-2 kinase and
immunological programming. _Front. Immunol_. 8, 1719 (2017). PubMed PubMed Central Google Scholar * Wek, R. C. & Staschke, K. A. How do tumours adapt to nutrient stress? _EMBO J_. 29,
1946–1947 (2010). CAS PubMed PubMed Central Google Scholar * Ye, J. et al. The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient
deprivation. _EMBO J_. 29, 2082–2096 (2010). CAS PubMed PubMed Central Google Scholar * Adam, I. et al. Upregulation of tryptophanyl-tRNA synthethase adapts human cancer cells to
nutritional stress caused by tryptophan degradation. _OncoImmunology_ 7, e1486353 (2018). PubMed PubMed Central Google Scholar * Loayza-Puch, F. et al. Tumour-specific proline
vulnerability uncovered by differential ribosome codon reading. _Nature_ 530, 490–494 (2016). ADS CAS PubMed Google Scholar * Bhushan, S. et al. α-Helical nascent polypeptide chains
visualized within distinct regions of the ribosomal exit tunnel. _Nat. Struct. Mol. Biol_. 17, 313–317 (2010). CAS PubMed Google Scholar * Woolhead, C. A., McCormick, P. J. & Johnson,
A. E. Nascent membrane and secretory proteins differ in FRET-detected folding far inside the ribosome and in their exposure to ribosomal proteins. _Cell_ 116, 725–736 (2004). CAS PubMed
Google Scholar * Caliskan, N. et al. Conditional switch between frameshifting regimes upon translation of dnaX mRNA. _Mol. Cell_ 66, 558–567 (2017). CAS PubMed Google Scholar *
Yelverton, E., Lindsley, D., Yamauchi, P. & Gallant, J. A. The function of a ribosomal frameshifting signal from human immunodeficiency virus-1 in _Escherichia coli_. _Mol. Microbiol_.
11, 303–313 (1994). CAS PubMed PubMed Central Google Scholar * Gurvich, O. L., Baranov, P. V., Gesteland, R. F. & Atkins, J. F. Expression levels influence ribosomal frameshifting at
the tandem rare arginine codons AGG_AGG and AGA_AGA in _Escherichia coli_. _J. Bacteriol_. 187, 4023–4032 (2005). CAS PubMed PubMed Central Google Scholar * Olubajo, B. & Taylor, E.
W. A. A −1 frameshift in the HIV-1 _env_ gene is enhanced by arginine deficiency via a hungry codon mechanism. _Mutat. Res_. 579, 125–132 (2005). CAS PubMed Google Scholar * Barak, Z.,
Lindsley, D. & Gallant, J. On the mechanism of leftward frameshifting at several hungry codons. _J. Mol. Biol_. 256, 676–684 (1996). CAS PubMed Google Scholar * Lainé, S., Thouard,
A., Komar, A. A. & Rossignol, J. M. Ribosome can resume the translation in both +1 or −1 frames after encountering an AGA cluster in _Escherichia coli_. _Gene_ 412, 95–101 (2008). PubMed
Google Scholar * Temperley, R., Richter, R., Dennerlein, S., Lightowlers, R. N. & Chrzanowska-Lightowlers, Z. M. Hungry codons promote frameshifting in human mitochondrial ribosomes.
_Science_ 327, 301 (2010). ADS CAS PubMed Google Scholar * Vredevoogd, D. W. et al. Augmenting immunotherapy impact by lowering tumor TNF cytotoxicity threshold. _Cell_ 178, 585–599
(2019). CAS PubMed Google Scholar * Zhou, F. Molecular mechanisms of IFN-γ to up-regulate MHC class I antigen processing and presentation. _Int. Rev. Immunol_. 28, 239–260 (2009). CAS
PubMed Google Scholar * Bourdetsky, D., Schmelzer, C. E. & Admon, A. The nature and extent of contributions by defective ribosome products to the HLA peptidome. _Proc. Natl Acad. Sci.
USA_ 111, E1591–E1599 (2014). ADS CAS PubMed PubMed Central Google Scholar * Yewdell, J. W. DRiPs solidify: progress in understanding endogenous MHC class I antigen processing. _Trends
Immunol_. 32, 548–558 (2011). CAS PubMed PubMed Central Google Scholar * Trentini, D. B. et al. Role for ribosome-associated quality control in sampling proteins for MHC class I-mediated
antigen presentation. _Proc. Natl Acad. Sci. USA_ 117, 4099–4108 (2020). CAS PubMed PubMed Central Google Scholar * Erhard, F. et al. Improved Ribo-seq enables identification of cryptic
translation events. _Nat. Methods_ 15, 363–366 (2018). CAS PubMed PubMed Central Google Scholar * Prasad, S., Starck, S. R. & Shastri, N. Presentation of cryptic peptides by MHC
class I is enhanced by inflammatory stimuli. _J. Immunol_. 197, 2981–2991 (2016). CAS PubMed Google Scholar * Starck, S. R. & Shastri, N. Nowhere to hide: unconventional translation
yields cryptic peptides for immune surveillance. _Immunol. Rev_. 272, 8–16 (2016). CAS PubMed PubMed Central Google Scholar * Chen, J. et al. Pervasive functional translation of
noncanonical human open reading frames. _Science_ 367, 1140–1146 (2020). ADS CAS PubMed PubMed Central Google Scholar * Chong, C. et al. Integrated proteogenomic deep sequencing and
analytics accurately identify non-canonical peptides in tumor immunopeptidomes. _Nat. Commun_. 11, 1293 (2020). ADS CAS PubMed PubMed Central Google Scholar * Laumont, C. M. &
Perreault, C. Exploiting non-canonical translation to identify new targets for T cell-based cancer immunotherapy. _Cell. Mol. Life Sci_. 75, 607–621 (2018). CAS PubMed Google Scholar *
Laumont, C. M. et al. Noncoding regions are the main source of targetable tumor-specific antigens. _Sci. Transl. Med_. 10, eaau5516 (2018). CAS PubMed Google Scholar * Pearson, H. et al.
MHC class I-associated peptides derive from selective regions of the human genome. _J. Clin. Invest_. 126, 4690–4701 (2016). PubMed PubMed Central Google Scholar * Laumont, C. M. et al.
Global proteogenomic analysis of human MHC class I-associated peptides derived from non-canonical reading frames. _Nat. Commun_. 7, 10238 (2016). ADS CAS PubMed PubMed Central Google
Scholar * Saulquin, X. et al. +1 Frameshifting as a novel mechanism to generate a cryptic cytotoxic T lymphocyte epitope derived from human interleukin 10. _J. Exp. Med_. 195, 353–358
(2002). CAS PubMed PubMed Central Google Scholar * Dersh, D., Yewdell, J. W. & Wei, J. A SIINFEKL-based system to measure MHC class I antigen presentation efficiency and kinetics.
_Methods Mol. Biol_. 1988, 109–122 (2019). CAS PubMed PubMed Central Google Scholar * McCarthy, M. K. & Weinberg, J. B. The immunoproteasome and viral infection: a complex regulator
of inflammation. _Front. Microbiol_. 6, 21 (2015). PubMed PubMed Central Google Scholar * Goldberg, A. L., Cascio, P., Saric, T. & Rock, K. L. The importance of the proteasome and
subsequent proteolytic steps in the generation of antigenic peptides. _Mol. Immunol_. 39, 147–164 (2002). CAS PubMed Google Scholar * Kalaora, S. et al. Combined analysis of antigen
presentation and t-cell recognition reveals restricted immune responses in melanoma. _Cancer Discov_. 8, 1366–1375 (2018). CAS PubMed PubMed Central Google Scholar * Strønen, E. et al.
Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. _Science_ 352, 1337–1341 (2016). ADS PubMed Google Scholar * Ali, M. et al. Induction of
neoantigen-reactive T cells from healthy donors. _Nat. Protocols_ 14, 1926–1943 (2019). CAS PubMed Google Scholar * Dong, L., Li, P., Oenema, T., McClurkan, C. L. & Koelle, D. M.
Public TCR use by herpes simplex virus-2-specific human CD8 CTLs. _J. Immunol_. 184, 3063–3071 (2010). CAS PubMed Google Scholar * Martin, M. Cutadapt removes adapter sequences from
high-throughput sequencing reads. _EMBnet.journal_ 17, 10–12 (2011). Google Scholar * Frankish, A. et al. GENCODE reference annotation for the human and mouse genomes. _Nucleic Acids Res_.
47, D766–D773 (2019). CAS PubMed Google Scholar * Langmead, B. Aligning short sequencing reads with Bowtie. _Curr. Protoc. Bioinformatics_ 32, 11.7.1–11.7.14 (2010). Google Scholar *
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. _Genome Biol_. 14, R36 (2013). PubMed PubMed Central Google Scholar
* Lauria, F. et al. riboWaltz: optimization of ribosome P-site positioning in ribosome profiling data. _PLoS Comput. Biol_. 14, e1006169 (2018). PubMed PubMed Central Google Scholar *
Li, H. et al. The Sequence Alignment/Map format and SAMtools. _Bioinformatics_ 25, 2078–2079 (2009). PubMed PubMed Central Google Scholar * Anders, S., Pyl, P. T. & Huber, W. HTSeq—a
Python framework to work with high-throughput sequencing data. _Bioinformatics_ 31, 166–169 (2015). CAS PubMed Google Scholar * Anders, S. & Huber, W. Differential expression analysis
for sequence count data. _Genome Biol_. 11, R106 (2010). CAS PubMed PubMed Central Google Scholar * Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for
comparing genomic features. _Bioinformatics_ 26, 841–842 (2010). CAS PubMed PubMed Central Google Scholar * Rainer, J., Gatto, L. & Weichenberger, C. X. ensembldb: an R package to
create and use Ensembl-based annotation resources. _Bioinformatics_ 35, 3151–3153 (2019). CAS PubMed PubMed Central Google Scholar * Jersie-Christensen, R. R., Sultan, A. & Olsen, J.
V. Simple and reproducible sample preparation for single-shot phosphoproteomics with high sensitivity. _Methods Mol. Biol_. 1355, 251–260 (2016). CAS PubMed Google Scholar * Tyanova, S.
et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. _Nat. Methods_ 13, 731–740 (2016). CAS PubMed Google Scholar * Ameziane, N. et al. A novel
Fanconi anaemia subtype associated with a dominant-negative mutation in RAD51. _Nat. Commun_. 6, 8829 (2015). ADS CAS PubMed Google Scholar * Zerbino, D. R. et al. Ensembl 2018. _Nucleic
Acids Res_. 46, D754–D761 (2018). CAS PubMed Google Scholar * Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and
proteome-wide protein quantification. _Nat. Biotechnol_. 26, 1367–1372 (2008). CAS PubMed Google Scholar * UniProt Consortium. UniProt: a worldwide hub of protein knowledge. _Nucleic
Acids Res_. 47, D506–D515 (2019). Google Scholar * Zhang, X. et al. Proteome-wide identification of ubiquitin interactions using UbIA-MS. _Nat. Protocols_ 13, 530–550 (2018). CAS PubMed
Google Scholar * Kalaora, S. et al. Use of HLA peptidomics and whole exome sequencing to identify human immunogenic neo-antigens. _Oncotarget_ 7, 5110–5117 (2016). PubMed PubMed Central
Google Scholar * Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. _Bioinformatics_ 28, 3150–3152 (2012). CAS PubMed
PubMed Central Google Scholar * Jurtz, V. et al. NetMHCpan-4.0: improved peptide–MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. _J.
Immunol_. 199, 3360–3368 (2017). CAS PubMed Google Scholar * Van der Auwera, G. A. et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices
pipeline. _Curr. Protoc. Bioinformatics_ 43, 11.10.1–11.10.33 (2013). Google Scholar * Li, H. D., Funk, C. C. & Price, N. D. iREAD: a tool for intron retention detection from RNA-seq
data. _BMC Genomics_ 21, 128 (2020). CAS PubMed PubMed Central Google Scholar * Andreatta, M., Alvarez, B. & Nielsen, M. GibbsCluster: unsupervised clustering and alignment of
peptide sequences. _Nucleic Acids Res_. 45, W458–W463 (2017). CAS PubMed PubMed Central Google Scholar * Vita, R. et al. The Immune Epitope Database (IEDB): 2018 update. _Nucleic Acids
Res_. 47, D339–D343 (2019). CAS PubMed Google Scholar * Abelin, J. G. et al. Mass spectrometry profiling of HLA-associated peptidomes in mono-allelic cells enables more accurate epitope
prediction. _Immunity_ 46, 315–326 (2017). CAS PubMed PubMed Central Google Scholar * Sarkizova, S. et al. A large peptidome dataset improves HLA class I epitope prediction across most
of the human population. _Nat. Biotechnol_. 38, 199–209 (2020). CAS PubMed Google Scholar * Krokhin, O. V. & Spicer, V. Peptide retention standards and hydrophobicity indexes in
reversed-phase high-performance liquid chromatography of peptides. _Anal. Chem_. 81, 9522–9530 (2009). CAS PubMed Google Scholar * Gatto, L. & Lilley, K. S. MSnbase-an R/Bioconductor
package for isobaric tagged mass spectrometry data visualization, processing and quantitation. _Bioinformatics_ 28, 288–289 (2012). CAS PubMed Google Scholar * MacLean, B. et al. Skyline:
an open source document editor for creating and analyzing targeted proteomics experiments. _Bioinformatics_ 26, 966–968 (2010). CAS PubMed PubMed Central Google Scholar * Mészáros, B.,
Erdos, G. & Dosztányi, Z. IUPred2A: context-dependent prediction of protein disorder as a function of redox state and protein binding. _Nucleic Acids Res_. 46, W329–W337 (2018). PubMed
PubMed Central Google Scholar * Ingolia, N. T., Brar, G. A., Rouskin, S., McGeachy, A. M. & Weissman, J. S. The ribosome profiling strategy for monitoring translation in vivo by deep
sequencing of ribosome-protected mRNA fragments. _Nat. Protocols_ 7, 1534–1550 (2012). CAS PubMed Google Scholar * Toebes, M. et al. Design and use of conditional MHC class I ligands.
_Nat. Med_. 12, 246–251 (2006). CAS PubMed Google Scholar * Hadrup, S. R. et al. Parallel detection of antigen-specific T-cell responses by multidimensional encoding of MHC multimers.
_Nat. Methods_ 6, 520–526 (2009). CAS PubMed Google Scholar * Philips, D., van den Braber, M., Schumacher, T. N. & Kvistborg, P. pMHC multiplexing strategy to detect high numbers of T
cell responses in parallel. _Methods Mol. Biol_. 1514, 93–101 (2017). CAS PubMed Google Scholar * Perez-Riverol, Y. et al. The PRIDE database and related tools and resources in 2019:
improving support for quantification data. _Nucleic Acids Res_. 47, D442–D450 (2019). CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS R.A. is supported by the Dutch Cancer
Society (KWF projects 10315, 11037 and 11574), the European Research Council (ERC–PoC 665317 and ERC-AdG 832844) and the Dutch science organization (NWO-TOP 91216002).Y.S. is supported by
the Israel Science Foundation grant no. 696/17, the ERC under the European Union’s Horizon 2020 research and innovation programme (grant agreement no. 770854), MRA (622106), Israel Science
Foundation (696/17), Rising Tide Foundation, Henry Chanoch Krenter Institute for Biomedical Imaging and Genomics, Estate of Alice Schwarz-Gardos, Estate of John Hunter, Knell Family and the
Hamburger Family. A.P. is supported by a long-term EMBO fellowship grant (EMBO ALTF 796-2018). O.B.B. and M. Alterlaar are supported by the Dutch NWO X-omics Initiative. J.O. and M.L. are
supported by the South-Eastern Regional Health Authority Norway, the Research Council of Norway, the Norwegian Cancer Society, Stiftelsen Kristian Gerhard Jebsen, the University of Oslo and
Oslo University Hospital. We thank M. Delic-Sarac, S. Meyer and T. J. Gjerdingen for HLA typing and processing of blood from in-house healthy donors; P. Kvistborg for providing selected
tetramers; and S. Reich-Zeliger, A. Nachshon, L. Eisenbach, A. Navon, S. Pinto, A. Peri, S. Cohen, A. Admon, A. Kacen and all members of the Agami laboratory for discussions. AUTHOR
INFORMATION Author notes * These authors contributed equally: Osnat Bartok, Abhijeet Pataskar, Remco Nagel AUTHORS AND AFFILIATIONS * Department of Molecular Cell Biology, Weizmann Institute
of Science, Rehovot, Israel Osnat Bartok, Eden Goldfarb, Deborah Hayoun, Ronen Levy, Michal Alon & Yardena Samuels * Division of Oncogenomics, Oncode Institute, The Netherlands Cancer
Institute, Amsterdam, The Netherlands Abhijeet Pataskar, Remco Nagel, Pierre-Rene Körner, Inger Z. M. Kreuger, Julien Champagne & Reuven Agami * Department of Cancer Immunology,
Institute for Cancer Research, Oslo University Hospital Radiumhospitalet, Oslo, Norway Maarja Laos, Weiwen Yang, Morten M. Nielsen & Johanna Olweus * Institute of Clinical Medicine,
University of Oslo, Oslo, Norway Maarja Laos, Weiwen Yang, Morten M. Nielsen & Johanna Olweus * Biomolecular Mass Spectrometry and Proteomics, Bijvoet Center for Biomolecular Research,
Utrecht Institute for Pharmaceutical Sciences, Utrecht University and Netherlands Proteomics Centre, Utrecht, The Netherlands Esther A. Zaal, Maarten Altelaar & Celia R. Berkers *
Department of Biochemistry and Cell Biology, Faculty of Veterinary Medicine, Utrecht University, Utrecht, The Netherlands Esther A. Zaal & Celia R. Berkers * Proteomics Facility, The
Netherlands Cancer Institute, Amsterdam, The Netherlands Onno B. Bleijerveld & Maarten Altelaar * Division of Molecular Oncology and Immunology, Oncode Institute, The Netherlands Cancer
Institute, Amsterdam, The Netherlands Xinyao Huang, Juliana Kenski & Daniel S. Peeper * Department of Surgical Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX,
USA Jennifer Wargo * Department of Genomic Medicine, The University of Texas MD Anderson Cancer Center, Houston, TX, USA Jennifer Wargo * Life Sciences Core Facilities, Weizmann Institute of
Science, Rehovot, Israel Alexander Brandis * The Nancy and Stephen Grand Israel National Center for Personalized Medicine, Weizmann Institute of Science, Rehovot, Israel Yishai Levin *
Department of Molecular Genetics, Weizmann Institute of Science, Rehovot, Israel Orel Mizrahi & Noam Stern-Ginossar * Department of Biological Regulation, Weizmann Institute of Science,
Rehovot, Israel Sacha Lebon * Department of Human Molecular Genetics and Biochemistry, Sackler School of Medicine, Tel Aviv University, Tel Aviv, Israel Tamar Geiger * Erasmus MC, Rotterdam
University, Rotterdam, The Netherlands Reuven Agami Authors * Osnat Bartok View author publications You can also search for this author inPubMed Google Scholar * Abhijeet Pataskar View
author publications You can also search for this author inPubMed Google Scholar * Remco Nagel View author publications You can also search for this author inPubMed Google Scholar * Maarja
Laos View author publications You can also search for this author inPubMed Google Scholar * Eden Goldfarb View author publications You can also search for this author inPubMed Google Scholar
* Deborah Hayoun View author publications You can also search for this author inPubMed Google Scholar * Ronen Levy View author publications You can also search for this author inPubMed
Google Scholar * Pierre-Rene Körner View author publications You can also search for this author inPubMed Google Scholar * Inger Z. M. Kreuger View author publications You can also search
for this author inPubMed Google Scholar * Julien Champagne View author publications You can also search for this author inPubMed Google Scholar * Esther A. Zaal View author publications You
can also search for this author inPubMed Google Scholar * Onno B. Bleijerveld View author publications You can also search for this author inPubMed Google Scholar * Xinyao Huang View author
publications You can also search for this author inPubMed Google Scholar * Juliana Kenski View author publications You can also search for this author inPubMed Google Scholar * Jennifer
Wargo View author publications You can also search for this author inPubMed Google Scholar * Alexander Brandis View author publications You can also search for this author inPubMed Google
Scholar * Yishai Levin View author publications You can also search for this author inPubMed Google Scholar * Orel Mizrahi View author publications You can also search for this author
inPubMed Google Scholar * Michal Alon View author publications You can also search for this author inPubMed Google Scholar * Sacha Lebon View author publications You can also search for this
author inPubMed Google Scholar * Weiwen Yang View author publications You can also search for this author inPubMed Google Scholar * Morten M. Nielsen View author publications You can also
search for this author inPubMed Google Scholar * Noam Stern-Ginossar View author publications You can also search for this author inPubMed Google Scholar * Maarten Altelaar View author
publications You can also search for this author inPubMed Google Scholar * Celia R. Berkers View author publications You can also search for this author inPubMed Google Scholar * Tamar
Geiger View author publications You can also search for this author inPubMed Google Scholar * Daniel S. Peeper View author publications You can also search for this author inPubMed Google
Scholar * Johanna Olweus View author publications You can also search for this author inPubMed Google Scholar * Yardena Samuels View author publications You can also search for this author
inPubMed Google Scholar * Reuven Agami View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS O.B., A.P. and R.N., conceived the project, designed
and performed experiments, analysed data and wrote the manuscript; R.A. and Y.S. conceived the project, designed experiments, wrote the manuscript and supervised the project; J.O. and M.L.
designed experiments for aberrant peptide immunogenicity testing and wrote the manuscript; M. Alon assisted in the peptidomics analysis; M.L. performed immunogenicity testing of aberrant
peptides; W.Y. produced monomers; M.M.N. optimized combinatorial tetramer staining and contributed to HLA typing and blood processing of in-house healthy PBMC donors; R.L. generated the
frameshift sequence database for peptidomics data; M. Alon, D.H. and E.G. performed the bioinformatics analyses of the peptidomics data; T.G. and D.H. performed the correlation analysis
between the synthetic and endogenous spectra; S.L. assisted in peptide validation; P.-R.K. performed the diricore analysis of ribosome profiling data; I.Z.M.K. and S.L. performed cloning of
reporters, western blot and flow cytometry analyses; J.C. performed the tGFP western blot; E.A.Z., C.R.B. and A.B. performed the metabolomics analyses; O.B.B., M. Alterlaar and Y.L.
performed mass spectrometry; X.H., J.K. and D.S.P. provided reagents and technical assistance for the T cell co-culture assays; J.W. helped in generating melanoma cell lines; and O.M. and
N.S.-G. assisted in performing ribosome profiling. All authors read and approved the manuscript. CORRESPONDING AUTHORS Correspondence to Yardena Samuels or Reuven Agami. ETHICS DECLARATIONS
COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PEER REVIEW INFORMATION _Nature_ thanks Nahum Sonenberg, Petra Van Damme, Jonathan Yewdell and the
other, anonymous, reviewer(s) for their contribution to the peer review of this work. PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps
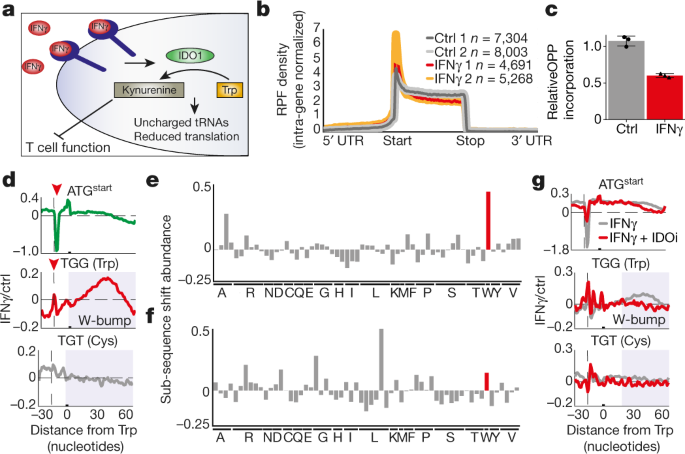
and institutional affiliations. EXTENDED DATA FIGURES AND TABLES EXTENDED DATA FIG. 1 DIRICORE ANALYSIS CHARACTERIZES RIBOSOME OCCUPANCY CHANGES AFTER IFNΓ AND TRYPTOPHAN-DEPLETION
TREATMENTS OF MELANOMA CELLS. A, Western blot analysis for IDO1 expression in control and IFNγ-treated conditions in three different melanoma cell lines as indicated (_n_ = 3). B, Tryptophan
(left) and kynurenine (right) levels as determined by mass spectrometry of the indicated melanoma cell lines in control conditions or 48h after the start of IFNγ treatment. Data represent
averages of three independent experiments ± s.d. C, Schematic depicting the principle behind diricore analysis: E, P and A-site occupancy of the ribosome can be mapped on a typical RPF at
9th, 12th and 15th positions, respectively. This allows a position-specific sub-sequence analysis to be performed (bottom left) to probe for the codon enriched differentially between two
conditions at E, P or A sites (presented as bar-plots). In addition, diricore analysis depicts 5′-RPF densities across a codon of interest (bottom right, presented as line-plots). D,
Metagene density profiles depicting global shifts of RPFs to the start of the coding sequence upon IFNγ treatment (red, yellow) as compared to control (grey). The _y_ axis is intra-gene
normalized RPF density. Ribosome profiling data of two independent biological replicates for MD55A3 (top) and 108T (bottom) are represented. E, Diricore analysis line plots depicting
differential ribosome occupancy (5′-RPF) at −30 to +60 codons in MD55A3 (left) and 108T (right) cells. Plots depict 5′-RPF densities after the start ATG codon (top, in green), tryptophan
codon (middle, in red) and cysteine codon (lower, in grey). The _y_ axis is the ratio between number of reads in IFNγ versus control conditions. F, Diricore analysis bar-plots depicting
differential codon usage (at position 15 of the RPFs) in IFNγ versus the control condition for MD55A3 (top) and 108T cells (bottom). Data represent the average of two independent biological
replicates. G, Diricore line plots depicting cumulative signal of RPFs across the coding region normalized into percentiles for ATF4, CDC6 and ATP5G1 in 12T control (grey tones) and
IFNγ-treated cells (yellow, red). W-bumps (greyed areas) indicate an increased number of reads downstream of the tryptophan codons (dashed lines). H, Tryptophan levels as measured by mass
spectrometry from 12T cells subjected to the indicated treatments. Bars represent the average of three independent experiments ± s.d. I, UCSC tracks representing IDO1 mRNA reads in MD55A3
cells in the indicated conditions as a measure for the induction of transcription of this gene. J, Metagene RPF density profiles for control (black), IFNγ (red), IDOi (grey) and IFNγ + IDOi
(green)-treated 12T cells. K, Diricore analysis bar plots depicting differential codon usage (at position 15 of the RPFs) in IFNγ- versus control-treated 12T cells. The graph represents the
average of two independent biological replicates. This experiment is the control of Fig. 1f. L, Tryptophan levels as measured by mass spectrometry in MD55A3 cells following 48-h growth in
control or tryptophan-depleted medium. Values represent the average of three independent replicates ± s.d. M, Metagene density profiles for 12T control (dark grey) and tryptophan depletion
cells (red). The lines represent the average of two independent biological replicates. N, Diricore analysis line plots depicting differential (IFNγ/control) ribosome occupancy (5′-RPF) at
−30 to +30 codons of the indicated amino acid in control versus tryptophan-depleted 12T cells. O, Diricore analysis bar plots depicting differential codon usage (at position 15 of the RPFs)
in control and tryptophan-depleted 12T cells. All diricore plots represent the average of at least two biological replicates. EXTENDED DATA FIG. 2 BUMP-FINDER IDENTIFIES
TRYPTOPHAN-ASSOCIATED BUMPS. A, Computational approach for unbiased detection of bumps in comparative ribosome profiling experiments. The algorithm scans transcripts in 100 windows of equal
length for peaks in ribosome occupancy and filters for differential peaks in treated versus untreated samples. B, C, Density of codons per amino acid in the region of 60 codons upstream and
downstream of the peak of bumps identified with bump-finder in ribosome profiling data of 12T cells. Data shown iare derived from two independent biological replicates treated with either
IFNγ (B) or control (C). D, Ratio between upstream and downstream reads 30 codons from the peak of bumps identified in control conditions when treated with IFNγ in MD55A3 (top) and 108T
cells (bottom). E, RPF density in control- (grey line) and IFNγ- (red line) treated conditions in MD55A3 and 108T cells. The area marked in grey indicates the W-bump region. F, Densities
(top lines) and heat maps (bottom) of ribosomal P-sites 100 nucleotides surrounding every tryptophan codon in control and IFNγ conditions in 12T, MD55A3 and 108T cells. G, Classification of
all transcripts containing tryptophan codons, one group associated with W-bumps (‘Bumps’), and another group that is not associated with bumps (‘No bumps’). Graphs represent RPF density in
the region of 300 nucleotides surrounding the tryptophan codon. Bumps indicated in grey shading. H, Bar plot depicting the enrichment of presence of two tryptophan codons within a region of
eight codons in the bumps group over the no bumps group. W indicates a codon for tryptophan, whereas X indicates all remaining amino acids. I, Heat map depicting frequencies of codons for
each amino-acid at every position 25 codons upstream and downstream, with respect to tryptophan. J, Line plots depicting RPF density at tryptophan stratified by distance between two
tryptophan residues (black). In each graph the control (the occurrence of a single tryptophan) is presented in red. EXTENDED DATA FIG. 3 PROTEOMICS ANALYSES AFTER IFNΓ TREATMENT OF MD55A3
MELANOMA CELLS. A, Volcano plot depicting overall changes in the proteome upon IFNγ treatment as observed by analysis of quantitative mass-spectrometry data. _x_ axis: log-transformed fold
change between IFNγ versus control conditions in MD55A3 cells; _y_ axis: corresponding log-transformed adjusted _P_ value calculated from three independent biological replicates. Highlighted
in blue are proteins that are significantly differentially expressed. Both IFNγ-mediated induction of IDO1 and WARS and the immunoproteasome components are indicated. B, Left, box plots
depicting log-transformed fold change in the levels of protein (red; average of three replicates) and mRNA (brown; average of two replicates) in IFNγ-versus control-treated cells. Proteins
were grouped according to the number of asparagine (N), tyrosine (Y) or phenylalanine (F) residues in the protein sequence. Boxes depict first, second and third quartiles; whiskers depict
the range excluding the outliers. Test: Wilcoxon test (two-tailed); NS, not significant, *_P_ < 0.05. For asparagine box plots the actual _P_ values are 0.7, 0.8, 0.055 for protein
quantification (left) and 0.8,0.2, 0.013 for RNA quantifications (right) in the order shown. Middle, same as left but for number of tyrosine (Y) residues. Actual _P_ values are 0.7, 0.86 and
0.02 for protein quantification (left) and 0.98, 0.13 and 0.14 for RNA quantifications (right) in the order shown. Right, same as middle but for number of phenylalanine (F) residues. Actual
_P_ values are 0.7, 0.97 and 0.23 for protein quantification (left) and 0.3, 0.015 and 0.0038 for RNA quantifications (right) in the order shown. C, Western blot analysis of ubiquitinylated
proteins in total cell lysates of MD55A3 cells mock- or IFNγ-treated, which were additionally incubated minus or plus MG132 (_n_ = 1). D, A panel showing proteins with increased abundance
in total cell lysates of MD55A3 cells treated with MG132 versus controls. Log2-transformed fold changes were calculated on data of three independent replicates. E, Box plots depicting log
fold change in protein levels (average of three replicates) in IFNγ-treated versus control conditions in MD55A3 cells treated with proteasome inhibitor, for proteins stratified for different
numbers of aspargine (N), tyrosine (Y) and phenylalanine (F) residues in their sequence. Boxes depict first, second and third quartiles; whiskers depict the range excluding the outliers.
Test: Wilcoxon test (two-tailed); NS, not significant and *_P_ < 0.05. Actual _P_ values are 0.34, 0.44, 0.77 and 0.96, 0.84, 0.024 and 0.29, 0.84, 0.34 in the order shown. F, Box plots
depicting protein length (number of amino acids) for stratified group of genes with increasing number of tryptophan (left) and asparagine (middle), with their ratios (right). G, Density of
RPFs (average of two replicates) 300 codons across individual tryptophan codons (black line), two tryptophans that are present within a distance of 8 codons (green line) and two tryptophans
that are present at a distance greater than 8 codons (red line). H, Box plot depicting bump scores (average of two replicates) for instances of two tryptophans separated by fewer than 8
codons (green) and more than 8 codons (red). Bump scores are calculated in MD55A3 cells. Boxes depict first, second and third quartiles; whiskers depict the range excluding the outliers.
Test: Wilcoxon Ttest (two-tailed); ***_P_ < 0.0005. Actual _P_ value is < 2.2 ×10−16. I, Box plot depicting protein level changes (log-transformed fold change, average of three
replicates) between IFNγ and control conditions. The graph represents genes that have two tryptophan codons within a distance of 8 codons (green) or genes having a distance of more than 8
codons between two tryptophans (red). Test: Wilcoxon test (two-tailed); ***_P_ < 0.0005. Actual _P_ value is < 2 ×10−16 J, Same as I, but for asparagine (N), in MD55A3 cells. Test:
Wilcoxon test (two-tailed); NS, not significant. Actual _P_ value is 0.83. EXTENDED DATA FIG. 4 REPORTER ASSAYS FOR THE DETECTION OF TRYPTOPHAN-ASSOCIATED OUT-OF-FRAME EVENTS. A, A
hypothetical model suggesting a possible mechanism causative for W-bumps. In the normal scenario, ribosomes do not encounter problems when translating a tryptophan and progress translation
at regular speed (top). Tryptophan shortage, on the other hand, can lead to stalling on the tryptophan codon (bottom left), or could in theory induce frameshifting events, leading to
aberrant peptide production (bottom right). As the secondary structure of growing polypeptide chains is attained in the lower tunnel of the ribosome, the loss of an α-helical secondary
structure in this tunnel could hamper ribosomal progression. B, Box plot depicting bump-score (from two replicates) in MD55A3 cells from the group of selected peptides with ordered
out-of-frame peptides (‘Selected’ from Fig. 3b) and every tryptophan in the proteome (‘All’ from Fig. 3b). Boxes depict first, second and third quartiles; whiskers depict the range excluding
the outliers. Test: two-sample _t_-test; *_P_ = 0.056. C, Schematic representation of the final protein sequences that would form due to frameshifting events. The in-frame construct (top)
contains a His tag and would end up in the pull-down (PD) fraction. Whenever a frameshift occurs at the position of the tryptophan in ATF4, this protein would lose its His tag, and
consequently would end up in the supernatant fraction (S). The +1 and +2 out-of-frame constructs (bottom) do not contain a His tag, whereby the resulting proteins always end up in the
supernatant fraction (S) in a His-tag pull-down assay. When frameshifting events take place, however, the His tag is incorporated into the peptide, whereby the resulting protein ends up in
the pull-down fraction (PD). D, Western blot analysis of a sequential V5-tag immunoprecipitation on the supernatant samples of the Frame reporter expressing cells from Fig. 3e (_n_ = 1). The
image indicates the presence of V5-tag-containing peptides that do not contain a His tag generated from the in-frame reporter. E, Tryptophan levels in MD55A3 cells expressing the
V5-ATF4-His reporter constructs that were used in Fig. 3e and in D. Tryptophan levels were analysed by mass spectrometry after 48 h of treatment. Bars represent the average of three
independent replicates ± s.d. F, Tryptophan levels in MD55A3 cells expressing the V5-ATF4-His reporter constructs that were used in H. Tryptophan levels were analysed by mass spectrometry
after 24 and 48 h of treatment in triplicate. G, Western blot analysis showing V5-tagged peptides in pull-down samples of MD55A3 reporter cells that were mock-, IFNγ- or IFNγ + IDOi-treated
(_n_ = 2). H, Western blot analysis of V5-tagged proteins in pull-down samples of MD55A3 reporter cells that were either mock-treated or cultured in tryptophan-depleted medium (_n_ = 1). I,
Western blot analyses showing pull-down assays followed by V5 staining on western blot of MD55A3 cells expressing the original reporter constructs as depicted in Fig. 3d (Wt), or MD55A3
cells expressing the same reporters where the tryptophan codon was mutated to a codon for tyrosine (Y mut, _n_ = 1). J, Amino acid levels as determined by mass spectrometry from lysates of
Tyr-depleted cells (48 h) versus control cells. Bars represent the average of three independent replicates ± s.d. K, Western blot analysis showing V5-tagged peptides in pull-down samples of
MD55A3 reporter cells that were mock-treated or depleted of tyrosine for 48 h (_n_ = 1). L, Flow cytometry analyses showing the histograms obtained of MD55A3 cells expressing the
V5-ATF4-tGFP reporters in all 3 frames in mock- and IFNγ-treated conditions. Plots are a representative graph out of a triplicate biological experiment. M, Flow cytometry analyses showing
the quantification of histograms obtained in L. Bars represent the average of three independent replicates ± s.d., *_P_ values in order from left to right: 4.0 × 10−6 and 9.5 × 10−4 as
determined by a two-sided _t_-test. N, Anti-V5-tag and anti-tGFP western blot analysis of whole cell lysates from MD55A3 cells expressing the indicated reporters, which were subjected to
mock or IFNγ treatments. In each blot the position of the full-length in-frame protein and the shorter out-of-frame protein are marked by the arrowheads (_n_ = 2).O, Western blot analyses
with anti-V5 antibody of His-tag pull-down samples of 888-Mel and D10 cells expressing the in-frame and +1 reporters (_n_ = 2). The cells were either grown in isolation (−), or co-cultured
with MART-1 specific T cells for 16 h (+) before the pull-down was performed. P, Western blot analysis showing IDO1 upregulation in 888-Mel and D10 cells in control and T cell co-culture
conditions (_n_ = 2). The same cells were used for pull-down experiments in Fig. 3m. Q, His-tag pull-down was executed on the lysates of mock and IFNγ-treated 888-Mel and D10 cells
expressing the in-frame and _+_1 reporters (_n_ = 2). Both supernatant (S) and His-tag pull-down samples (PD) of these cells were stained with V5 antibodies. R, The lysates of cells used in
Q were used for a western blot analysis to show the level of IDO1 induction (_n_ = 1). S, Tryptophan levels as determined by mass spectrometry analysis of 888-Mel and D10 lysates in mock,
IFNγ-treated and tryptophan-depleted conditions. Bars represent the average of three independent replicates ± s.d., except for the first bar, which is an average of two independent
replicates. T, Western blot analyses with anti-V5 antibody of His-tag pull-down samples of 888-Mel and D10 that were mock-treated (+), or grown in tryptophan-less medium for 48 h (−) before
the pull-down was performed (_n_ = 2). EXTENDED DATA FIG. 5 PROTEOMICS ANALYSIS OF 2D-LC–MS/MS DATA REVEALS THE INDUCTION OF ENDOGENOUS ABERRANT PEPTIDES AFTER IFNΓ TREATMENT OF MD55A3
MELANOMA CELLS. A, PCA plot illustrating the clustering of 2D-LC-MS/MS data from MD55A3 cells that were treated with IFNγ for 48 h as compared to mock. Both conditions were treated with
MG-132 for the last 4 h. B, Gene set enrichment analysis (GSEA)-based depiction of the proteomics data showing the induction of the IFN response genes following IFNγ treatment of MD55A3
cells. C, Schematic representation of the in silico generation of +1 and −1 frameshifted protein database (Methods). D, Bar plot depicting number of hits for the detected aberrant
(frameshifted) polypeptides in the Ensemble translations database of coding and non-coding genes. Only the aberrant peptides without any matches (_n_ = 0) were retained as true hits (red
bar). E, Heat map showing frameshifted peptides identified in the full proteome of IFN-treated MD55A3 cells along with sequence information. F, Line plot depicting log-scaled fold change in
proteomic intensity values of IFNγ- versus mock-treated MD55A3 cells. G, Same as F, but for LFQ normalized intensity values of frameshifted peptides hits along with reverse peptide hits. H,
Same as F, but only for reverse peptide hits. I, Same as F, but only for frameshifted peptide hits. J, Gene set enrichment analysis (GSEA)-based depiction of the proteomics data showing the
induction of the HLA genes following IFNγ treatment of MD55A3 cells. K, GO term analysis on the differentially expressed proteins detected in J. L, Anti-V5-tag and anti-tGFP western blot
analysis of whole cell lysates from A375 cells expressing the indicated reporters, which were subjected to mock treatment or tryptophan depletion (_n_ = 2). In each blot the position of the
full-length in-frame protein and the shorter out-of-frame protein are marked by the arrowheads. M, FACS plots representing the signal for H-2Kb-bound SIINFEKL peptides in A375 cells
expressing H-2Kb in combination with in-frame (Frame) or +1 out-of-frame (+1) V5-ATF41–63-tGFP-SIINFEKL reporters. Graphs shown are a representative curve from one of three independent
experiments. EXTENDED DATA FIG. 6 QUALITY CONTROL ANALYSIS FOR THE IMMUNOPEPTIDOMICS OF MD55A3 CELLS. A, Length distribution of the identified peptides. B, HLA binding prediction using
netMHCpan shows a high percentage of eluted peptides predicted to bind MD55A3 haplotypes. C, HLA‐I peptides were clustered by Gibbs clustering to assign the peptides to the different HLA
alleles of the patient. Logos were created to identify the HLA alleles’ motif using all peptides matching to this allele in the IEDB database (top). Peptides that were identified in the
cells treated with IFNγ, tryptophan-derived (mTRP) and non–treated (NT) were clustered to 1–6 clusters, and only the best fit (highest Kullbach Leiber distance) was used for comparison with
the IEDB. The clusters show the motifs expected of the cells’ HLA alleles. The last column indicates the outlier peptides that were not clustered. EXTENDED DATA FIG. 7 IMMUNOPEPTIDOMICS
ANALYSIS IDENTIFIES TRYPTOPHAN-ASSOCIATED ABERRANT PEPTIDES. A, Scheme summarizing the filtration steps that led to the detection of HLA-bound aberrant peptides, containing W-associated
frameshift sequences (derived from 16 samples: 4 NT, 4 +IFNγ, 4 mTpr, 4 metastases from patient 55); immunopeptidomics spectra were searched against W-specific frameshifted human proteome.
MS-identified peptides were filtered according to described steps, including HLA binding prediction, spectral quality and source gene expression validation in transcriptome or translatome
datasets. Following the above steps, 28 aberrant peptides were detected either in MD55 metastasis or treated MD55A3 samples, and not in the untreated cells. B, Predicted hydrophobicity index
(HI) and observed retention time (RT) of canonical peptides (black) and aberrant peptides at two filtration steps described in a, step 3 (blue) and step 7 (red). The observed correlation
between the RT versus HI, (using the SSRCalc tool31,70) supports the identification of the aberrant peptides presented in A. C, Schematic representation of aberrant peptides KCNK6 and PCNX2
detected via immunopeptidomics, derived from a −1 and +1 frameshift at a Trp codon (marked in red), respectively. The in-frame and out of frame amino acid sequences are indicated above
(black) and below (red/green) the mRNA sequence, respectively. D, Venn diagram including the 28 aberrant peptides detected following the filtration steps presented in A (were not identified
in the untreated MD55A3 cells), grouped according to the samples they were detected in. E, Mean LFQ normalized intensities. Canonical and aberrant peptides that exhibit above threefold
change in their intensities, are marked in black and blue/red, respectively. F, G, Similarities between the aberrant (_n_ = 28) and canonical (_n_ = 6,304) bound peptides. F, Andromeda
score. G, Binding affinity prediction with NetMHCpan showing a similar binding distribution of the canonical and aberrant peptides. The aberrant peptides are highlighted in red. Dashed line
mark strong binders rank cutoff (<0.5). H, A list of eight out of the thirteen −1 and +1 out-of-frame and _trans_-frame peptides found in the metastasis samples of patient 55. The colours
of the peptide sequences represent in-frame (black), +1 frame (green) and −1 frame (red). EXTENDED DATA FIG. 8 TANDEM MASS SPECTRA OF ENDOGENOUS ABERRANT PEPTIDES IDENTIFIED IN PATIENT 55
MATCH THE SPECTRA IDENTIFIED IN METASTASIS SAMPLES FROM PATIENT 42. Left, experimental spectra of peptides identified in metastasis samples from patient 42, who shares three of six HLA-I
alleles with patient 55 (A03:01; B07:02; C07:02). Right, head-to-tail plots show similarities between overlapping ions of the two spectra, Top (blue), patient 42; bottom (red), patient 55
(MD55). EXTENDED DATA FIG. 9 IDENTIFICATION OF T CELLS REACTIVE TO TRYPTOPHAN-DERIVED ABERRANT PEPTIDES. A, Flow cytometry analysis of CD8+ T cells reactive to BV421 and PE-labelled pMHC
multimers complexed with the ZNF513-derived aberrant peptide in co-cultures of naïve CD8+ T cells and autologous monocyte-derived dendritic cells pulsed with peptide (right) or DMSO vehicle
(left). Cells were from an HLA-C*07:02pos healthy donor. B, Boolean gating strategy for analysis of combinatorial pMHC multimer staining limits false positive signals arising due to
background fluorescence when combining in one staining multiple different pMHC multimers labelled with different fluorochromes. In total, four different fluorochromes were used in pMHC
multimer preparation and combined into four dual-colour pMHC multimer pairs, each pair complexed with a different peptide. For simplicity, gating for only one dual-colour pMHC multimer
population is shown. Gating strategy: (1) Live CD8+ T cell singlets were identified with the use of FSC and SSC gates, live/dead fixable near-IR dead cell and CD8 staining. (2) Separate
gates were used to define positive events in each pMHC channel. (3) NOT gates were used to select CD8+/pMHC multimer− events for each pMHC channel. (4) AND gates for two pMHC multimer+
populations and NOT gates for each of the remaining pMHC channels were used to select for cells that are positive only in two channels. Events staining positively for one or more than two
pMHC channels were gated out. (5) AND gate was used to combine all pMHC NOT gates. (6) OR gate was made with gates from step 4 and 5. The final plot shows only CD8+ T cells that are positive
for two pMHC channels and excludes cells positive for only one or positive for more than two channels. C, For analysis of T cells upregulating the activation marker CD137, viable CD8+ T
cell singlets were identified by FSC and SSC gates, live/dead fixable near-IR dead cell staining and CD8 staining. Subsequently, activated CD8+ T cells were identified as CD137+ events, in
which cut-off was set based on staining of T cells incubated without target cells. Plots depicting CD137+ events show the KCNK6 T cell clone 1 incubated with target cells pulsed with the
KCNK6 aberrant peptide (1 μg ml−1, top), or not (bottom). SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION This file contains Supplementary Information File contains Supplementary
Methods, Supplementary Tables 1-3 and Supplementary Figures 1-4. REPORTING SUMMARY RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Bartok, O., Pataskar,
A., Nagel, R. _et al._ Anti-tumour immunity induces aberrant peptide presentation in melanoma. _Nature_ 590, 332–337 (2021). https://doi.org/10.1038/s41586-020-03054-1 Download citation *
Received: 09 January 2020 * Accepted: 30 October 2020 * Published: 16 December 2020 * Issue Date: 11 February 2021 * DOI: https://doi.org/10.1038/s41586-020-03054-1 SHARE THIS ARTICLE Anyone
you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by
the Springer Nature SharedIt content-sharing initiative