Identification of a locus conferring dominant resistance to maize rough dwarf disease in maize
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Maize rough dwarf disease (MRDD) is a severe viral disease of maize that occurs worldwide, particularly in the summer maize-growing areas in China, resulting in yield losses and
quality deterioration in susceptible maize varieties. An effective solution to control MRDD is to use resistance genes to improve the behavior of susceptible genotypes. Here, we employed
maize F2 populations derived from a cross between susceptible line S221 and resistant line K36 for the deep sequencing of the two DNA pools containing extremely resistant and susceptible F2
individuals, and used traditional linkage analysis to locate the resistance-related genomic region. The results showed that MRDD resistance in K36 was controlled by a single dominant locus,
and an associated region was identified within the genomic interval of 68,396,487 bp and 69,523,478 bp on chromosome 6. Two simple sequence repeat (SSR) markers 6F29R29 and 6F34R34 were
tightly linked to the MRDD resistance locus. The findings of the present study improve our understanding of the inheritance patterns of MRDD resistance, and should inform MRDD-resistant
maize breeding programs. SIMILAR CONTENT BEING VIEWED BY OTHERS DISCOVERY OF STRIPE RUST RESISTANCE WITH INCOMPLETE DOMINANCE IN WILD EMMER WHEAT USING BULKED SEGREGANT ANALYSIS SEQUENCING
Article Open access 17 August 2022 A TRANSCRIPTION FACTOR _ZMGLK36_ CONFERS BROAD RESISTANCE TO MAIZE ROUGH DWARF DISEASE IN CEREAL CROPS Article 14 September 2023 SEQUENCING
TRAIT-ASSOCIATED MUTATIONS TO CLONE WHEAT RUST-RESISTANCE GENE _YRNAM_ Article Open access 19 July 2023 INTRODUCTION Maize rough dwarf disease (MRDD) is a global viral disease in maize (_Zea
mays_ L.). It is particularly prevalent in the summer maize-growing areas in China and can lead to more than 30% reduction in yield, and can even result in 100% yield loss in severely
infected fields1,2,3. This disease was first detected in 1949 in Italy. Four members of the family Reoviridae belonging to the genus _Fijivirus_ are the causal pathogens of MRDD, and include
maize rough dwarf virus (MRDV), Mal de Rio Cuarto virus (MRCV), rice black streaked dwarf virus (RBSDV), and southern rice black streaked dwarf virus (SRBSDV). However, the virus strains
vary between different regions. MRDV affects maize in Europe, MRCV in South America, and RBSDV and SRBSDV in East Asia4,5,6,7,8. These closely related viruses are transmitted in a persistent
manner by planthoppers (_Laodelphax striatellus_) that infect common plant hosts such as rice, wheat, and maize. In China, RBSDV and SRBSDV are the pathogens of MRDD that can be transmitted
from infected wheat to maize by planthoppers6. MRDD is particularly severe in the Huang-Huai-Hai River summer maize-growing region when maize is sown around the wheat harvesting period1.
MRDD symptoms typically include dwarfed plant height, dark-green leaves that appear rough due to waxy enations, malformed tassels and upper leaves, and small ears or heading failure. In
China, the prevalence of MRDD has continued to increase with global warming and the reduced use of pesticides, which significantly reduces yield and results in quality deterioration in
maize2,3. To date, the use of resistant maize varieties remains the most effective and economical way to control this disease. Thus, mapping and cloning genes underlying the Quantitative
Trait Loci (QTLs) conferring resistance to MRDD are necessary for the development of resistant hybrids. In recent years, a few studies focusing on resistant germplasm screening under natural
infection conditions have been conducted in China9,10,11,12,13. These investigations suggested that there is a lack of germplasm resources that are immune to MRDD, but a limited number of
highly resistant lines were identified in different environments, and were mainly derived from a US hybrid, P78599. However, significant differences in resistance among various germplasm
resources have been reported9,10,13,14. Previous studies have indicated that the inheritance of MRDD resistance in maize is quantitative in nature14,15,16. Several QTLs have been identified
using F2:3 segregating populations and recombined inbred lines (RIL) derived from crosses between susceptible and resistant lines. The loci of the identified QTLs from the different
resistant lines vary2,17,18,19,20,21,22,23. However, some QTLs confer similar genetic effects in resistance among different maize inbred lines, such as the major QTL on chromosome 8
(binn8.03) that is associated with a 24.2–39.3% decrease in disease severity and is inherited in a recessive manner22. Although a number of resistant QTLs have been identified, the majority
are derived from the US hybrid P78599, including Qi 319 and X17824,25. Therefore, the identified QTLs may represent only a small portion of the available genetic resistance to MRDD in maize.
Genome-wide association studies (GWAS) allow for the detection of natural variations in complex genetic traits at a relatively high resolution in plants26,27,28. Recent studies have
employed GWAS to detect MRDD resistance-related loci in maize using single-nucleotide polymorphism (SNP) markers covering the entire maize genome25,29,30. Similar to the findings of linkage
analysis, GWAS indicated that each chromosome has a locus/loci associated with MRDD resistance. However, no MRDD resistance-related locus has been successfully cloned from maize to date. To
identify the genes/loci that confer resistance to MRDD, the present study employed a novel germplasm resource that exhibits MRDD resistance. The combined deep sequencing of two DNA pools
containing extremely resistant or susceptible F2 individuals and traditional linkage analysis using F2 populations derived from a cross between a maize susceptible inbred line, S221, and a
maize resistant inbred line, K36, were conducted. A genomic region associated with dominantly inherited MRDD resistance was identified on chromosome 6. Our results improve the understanding
of MRDD resistance inheritance in maize and should inform the development of breeding schemes for MRDD-resistant maize lines. RESULTS EVALUATION AND GENETIC ANALYSIS OF MRDD RESISTANCE The
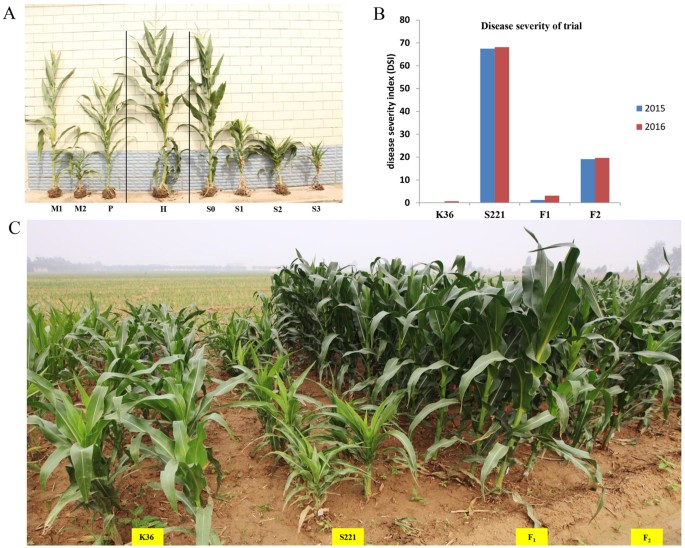
parental lines, S221 and K36, and their F1 and F2 progenies were evaluated for MRDD resistance in 2015 and 2016 (Fig. 1A,B). Obvious symptoms of MRDD were observed in the field trial under
naturally infested conditions in both 2015 and 2016 at Shijiazhuang; a summer maize-growing region in China. The average disease severity index (DSI) scores of K36 and S221 were 0.32 and
67.82 in the two years, respectively (Fig. 1C and Supplementary Table S1). In the two F2 populations planted in 2015 and 2016, respectively, there were more plants with disease scores of 0
or 3 than with scores of 1 or 2 (phenotyping section in Method & Supplementary Table S1). The disease score distributions were similar between the two F2 populations. The DSI in F1
ranged from 1.25 to 3.13, which is similar to that of the paternal line, K36, which is highly resistant to MRDD, thereby suggesting that the MRDD resistance in F1 was derived from K36 in a
dominant inheritance pattern (Supplementary Table S1). Meanwhile, resistance segregated in the two F2 populations with similar disease incidences of 28.3% and 27.8%, and DSIs of 19.1 and
19.6 in both years (Supplementary Table S1). To characterize the inheritance pattern of MRDD resistance, the disease resistance scores of the F1 and F2 individuals were statistically
analyzed. The F1 plants showed resistance to MRDD, while the F2 populations exhibited variation in disease resistance scores. When the F2 plants were classified into two groups, namely,
resistant or susceptible, the chi-square test indicated that the segregation ratio of the resistant and susceptible plants fitted the expected 3:1 segregation ratio (χ2 = 2.917, 3.835,
respectively), suggesting that MRDD resistance from K36 was controlled by a single dominant locus (Table 1). VIRUS DETECTION MRDD was detected in the maize plants, including the susceptible
parent S221-S and the individuals making up the S-pool with dwarf symptoms, but was not detected in the resistant parent, K36, and the 30 plants making up the R-pool (Fig. 2). The real-time
(RT)-PCR amplified fragment within the expected size (568 bp) from the cDNA of plants with dwarf symptoms was sequenced. Comparison of the obtained sequences with those deposited in GenBank
showed that the sequenced fragment had 94% identity with that of the RBSDV from Wuhan (AJ291706), Jiangsu (KC875238, KM921681), Shandong (JX421771), Anhui (KX660762), China, and 93% identity
with the RBSDV from Baoding (DQ407917), China (Supplementary Fig. S1). Additionally, two F2 populations from the same cross (S221 × K36) were planted as controls without virus infection.
One population consisting of 180 individuals was planted on September 15, 2016 in a protected greenhouse. The other population for the RIL population construction consisting of 846
individuals was sowed on June 20, 2016 to avoid planthopper infection. The plants from both populations showed no dwarf symptoms caused by RBSDV. Together with the high homology between the
sequences obtained from samples exhibiting MRDD symptoms and the RBSDV sequences from the maize planting zone of China from GenBank, the results suggested that the dwarf symptoms in the
parental varieties and F2 individuals were caused by RBSDV infection. HIGH-THROUGHPUT SEQUENCING ANALYSIS Based on the phenotypic screening for MRDD resistance, four types were classified
according to disease severity scores ranging from 0 to 3. From the F2 population, two contrasting DNA bulks were constructed by pooling equal volumes of DNA from R plants (disease severity
score 0) and S plants (disease severity score 3) to form R and S bulks, respectively. Specific-locus amplified fragment sequencing (SLAF-seq) for the two BSA bulks was conducted, resulting
in 12.97 Mb fragments from the two parents and 19.48 Mb fragments from the two bulks after exclusion of the low-quality fragments in the constructed SLAF library (Table 2). When the obtained
SLAF-seqs were matched to the reference genome, the results showed that over 92% of the reads could be mapped to the reference genome, and a total of 182,132 high-quality SLAF-tags were
developed with an average sequencing depth of 24.89-fold in the parental lines and 32.73-fold in the F2 bulks. Approximately 22,644 of the 182,132 SLAF-tags were polymorphic. A total of
709,670 SNPs were detected with ~4% and ~19% heterozygosity in the parental lines and F2 bulk pools, respectively (Table 2). Based on the results of the SLAF/SNP marker positioning on the
genome, the number of markers on each chromosome were calculated (Supplementary Table S2). Of these markers, a total of 181,235 SLAFs and 707,448 SNPs were localized to specific chromosomes.
The distribution diagrams for the SLAF/SNP markers on each chromosome were drawn based on their position on the genome (Fig. 3). The markers were distributed equally on each chromosome, and
the maize genome was successfully simplified by using this restriction site associated DNA sequencing approach. ASSOCIATION ANALYSIS The segregation ratio of resistance to MRDD in the F2
population indicated that the trait is controlled by a dominant locus. Thus, the association threshold of the ΔSNP-index value is expected to be 0.67. By examining the ΔSNP-index plot, peak
regions above the threshold value were defined as association regions. After calculations and fitting analysis, the ΔSNP indices of the two bulks were obtained (Fig. 4A). No region exceeded
the theoretical threshold on each chromosome, although a region on chromosome 6 was found near the theoretical threshold (Fig. 4B). When the threshold was reduced to 0.34 (quantile 99 of the
ΔSNP-index), a possible trait-related candidate region was obtained, which spanned 12.99 Mb (61,960,714 to 74,957,430 bp) on chromosome 6 of the reference genome and harbored 374 genes. The
Euclidean distance (ED) method was employed to further explore the trait-related region. Initially, 158,151 high-quality SNPs were obtained from a total of 709,670 SNPs by filtering out
551,519 SNP sites with read support less than 4 in the two pools. Then, a total of 59,665 different SNPs were identified from the obtained high-quality SNPs. Finally, after the ED value was
calculated, the correlation values were obtained using the local linear regression LOESS method (Fig. 4B). A possible trait-related candidate region was obtained when the threshold was
0.458. This associated region spanned 13.80 Mb from 55,724,783 to 69,523,478 bp on chromosome 6 of the reference genomic sequence, where the candidate region obtained using ΔSNP-index method
was also mostly covered. SSR ANALYSIS To validate the MRDD resistance-associated genomic region and to investigate recombination among the F2 individuals, SSR markers in the target region
were selected for linkage analysis. First, 35 SSRs distributed in the associated region were used in the assessment of the two parents. Of these, 11 markers showed polymorphism between the
two parents. After 126 individual plants from the F2 population were genotyped with the 11 SSR markers, a short linkage map was constructed using the JoinMap 4 program. The 11 markers
covered a genetic distance of 4.6 cM on chromosome 6. The major MRDD resistance locus on chromosome 6 was mapped between the 6F29R29 and 6F34R34 markers, which were 0.1 cM apart (Fig. 4C).
According to the genomic sequence of chromosome 6 in the B73 genome assembly, the physical position of 6F29R29 starts from 68,396,487 bp, whereas 6F34R34 starts from 73,915,399 bp. The two
markers covered approximately 5.52 Mb of the candidate region (Fig. 4C). A summary of the results from both the association analysis of the SLAF-seq and SSR analysis indicated that an
MRDD-resistance locus resides on chromosome 6. The candidate locus was designated as Rmrdd6 (resistance to maize rough dwarf disease on chromosome 6), and is located at the intersection of
the three obtained candidate regions, with the physical positions starting from 68,396,487 bp to 69,523,478 bp, encompassing approximately 1.13 Mb, and harboring 32 candidate genes.
CANDIDATE GENE IDENTIFICATION To identify candidate genes in the MRDD resistance-associated region, the initial SNPs within the exonic region between the two parents were analyzed and
variations were annotated (Supplementary Table S3). A total of 32 genes were included in the associated region. Gene ontology (GO)-based gene functional enrichment analysis was performed,
and the 32 genes were characterized according to cellular component, molecular function, and biological process. The results revealed that the most enriched terms were in the category of
cellular component, such as integral to cell wall (GO:0005618), anchored component of plasma membrane (GO:0046658), chloroplast envelope (GO:0009941), glycosylphosphatidylinositol (GPI)
transamidase complex (GO:0042765), and chloroplast (GO:0009507). Of the 32 genes, 9 possess known or hypothetical functions, and 23 genes encode uncharacterized proteins based on GO analysis
and the publicly available B73 annotated genome (http://plants.ensembl.org/Zea_mays/Info/Index). Among the 9 annotated genes, GRMZM2G161673 is predicted to encode magnesium protoporphyrin
IX methyltransferase and has methyltransferase activity in chloroplasts. GRMZM2G471321, also known as the _Tangled1_ gene, is required for the spatial control of cytoskeletal arrays that are
associated with cell division during maize leaf development31. GRMZM2G156422 encodes a transmembrane protein that is essential for retrograde transport in the trans-Golgi network. The
NAC-like gene GRMZM2G091490 is predicted to encode a transcription factor that plays an important role in the regulation of plant growth and development, hormone regulation, and responses to
various stresses32,33. Another NAC-like gene GRMZM2G110983 is predicted to encode a protein containing a ubiquitin-conjugating enzyme E2 catalytic (UBCc) domain that is involved in a
ubiquitin-mediated protein degradation pathway34. GRMZM2G478892 encodes a protein belonging to the O-glycosyl hydrolase family 17 and hydrolyzes O-glycosyl compounds in the cell wall matrix
of plants, and also has diverse roles in plant defense and plant development35. GRMZM2G175867, a member of the DEAD-box ATP-dependent RNA helicase family, plays important roles in RNA
metabolism and abiotic stress responses in chloroplasts and mitochondria36. GRMZM2G178602 encodes a glycosylphosphatidylinositol (GPI) transamidase component Gpi16 subunit family protein
that is involved in anchor biosynthesis, acting as an anchor for the attachment of cell surface proteins to the cell membrane37. GRMZM2G178616 encodes phosducin-like protein 3 that plays a
central role in cell division in the microtubule assembly pathway38. Furthermore, 7 SNPs with nonsynonymous mutations were identified in this genomic region, corresponding to 5 genes that
might be related to MRDD resistance. In particular, GRMZM2G384564 is more interesting because it had 3 SNPs with nonsynonymous mutations in the paternal line K36. DISCUSSION ACCURATE
PHENOTYPIC EVALUATION Accurate phenotypic evaluation is critical for marker-trait association analyses39. As with all viral diseases, MRDD requires interactions among an effective vector,
the virulent pathogen, a susceptible host, and an appropriate environment40. Wheat is one of the preferred hosts for the planthopper insect vector (_L_. _striatellus_) because it provides an
optimum environment for the insects to complete their development, after which they then migrate to adjacent maize crops. In the present study, natural infection was used because the virus
causing MRDD is transmitted in a persistent propagative manner, and the prevalence of MRDD in the experiment was attributable to the extensive sowing of wheat prior to that of maize. Virus
detection showed that the dwarf symptoms in the infected plants were caused by MRDV. Therefore, natural infection ensures accurate phenotypic evaluation. In 2015 and 2016, 98% of the plants
of maternal line S221 were infected, which is indicative of relatively stable and uniform infection conditions and implies that the results of the MRDD resistance evaluation were appropriate
for further marker-trait association analysis. Our study further proved that natural infection is a suitable method for evaluating MRDD resistance17,22,23. GENETIC ANALYSIS OF MRDD
RESISTANCE In the present study, the paternal line K36 displayed consistent resistance, whereas the maternal line S221 showed susceptibility. Although there are several infected plants in
their F1 populations, the majority, like K36, exhibited MRDD resistance. The F2 populations in the genetic analysis, consisting of an appropriate number of individuals, ensured that the
results of the chi-square test were accurate. The segregation ratio of resistant and susceptible plants of the F2 population was 3:1, indicating that a single dominant gene controlled the
resistance trait from K36. Our results differ from those of previous reports whereby MRDD resistance in maize was found to be controlled by several genes, each with a small effect14,15,16.
Different resistant maize germplasm resources display variable resistance to MRDD9,10,11,12,13. Several characterized highly resistant inbred lines, including Qi319 and X178, derived their
resistance from the US hybrid P78599, and consequently, several linkage-based mapping studies in China identified QTLs that control resistance2,21,22. The identification of a dominant gene
in K36 in this study broadens the genetic basis and enriches our understanding of the genetic mechanism underlying differences in MRDD resistance. EFFECTIVENESS OF THE SLAF-SEQ ANALYSIS AND
IDENTIFICATION OF RESISTANCE-ASSOCIATED REGIONS Traditional gene mapping and map-based cloning methods have facilitated the identification of molecular markers that flank and co-segregate
with a specific locus41. BSA is an efficient method for the rapid identification of molecular markers linked to the target gene or genomic region in two bulks showing clear phenotypic
differentiation42. However, the availability of DNA markers is the main factor limiting BSA effectiveness43. Next-generation sequencing technologies provide a comprehensive and
cost-effective means of unraveling the genetic diversity of genomes for accelerating gene mapping and isolation. The combination of SLAF-seq and BSA circumvents the limitation of DNA marker
availability and does not require complete genotyping. This strategy has been successfully applied to various plant species including wheat44, rice43,45, sorghum46, and sunflower47. In the
present study, both BSA-SLAF-seq and linkage analysis were conducted to detect resistance-related genomic regions. More than 182,132 SLAFs were developed and 709,670 SNP markers were
identified, all of which were of high quantity and quality. The SLAFs and SNPs were distributed across each chromosome. The integrity and accuracy of these markers are relatively high
compared to the 54,788 and 56,635 SLAFs developed by Yu and Xia when they identified genes associated with defective, pale green bundle sheaths and inflorescence meristems in maize,
respectively48,49. Therefore, the obtained markers provided sufficient data for the identification of candidate MRDD resistance-associated regions in this study. SLAF-seq analysis identified
one resistance-associated genomic region on chromosome 6 encompassing 61,960,714 bp to 69,523,478 bp. Subsequently, SSR markers were selected for linkage analysis to validate the identified
region. The linkage results further confirmed the associated genomic region on chromosome 6 and narrowed the region down to 1.13 Mb, encompassing the genomic region from 68,396,487 bp to
69,523,478 bp. Previous studies have identified numerous QTLs conferring resistance to MRDD through linkage mapping or genome-wide association analysis. Additionally, a few of these
identified QTLs were consistently associated with MRDD in various studies, such as a major QTL locus on chromosome bin 8.032,18,22,23. Although some QTLs were also identified on chromosome
617,25,30,50,51, these did not overlap with the genomic region identified in the present study. Therefore, our findings provide new insights into the genetic architecture of MRDD resistance
in maize. The identification of markers that are significantly associated with traits of interest is mainly used for MAS in plant breeding programs. In the present study, the Rmrdd6 that
confers resistance to MRDD was mapped to a 1.13-Mb interval. Two SSR markers, 6F29R29 and 6F34R34, were tightly linked to the MRDD resistance gene, which can be efficiently used in the MAS
of MRDD resistance and accelerate the improvement of maize breeding. POTENTIAL CANDIDATE GENES RESISTANT TO MRDD The ultimate goal of gene mapping is to obtain the target candidate gene. In
this study, the GRMZM2G384564 gene was of great interest to MRDD resistance, as three non-synonymous mutations were identified in the paternal line K36 when compared to reference B73 and
maternal line S221. Whether these mutations conferred resistance to MRDD in K36 requires further validation. GRMZM2G384564 is a maize-specific gene, but its function remains unclear.
Therefore, further assessment of this gene is warranted. Dwarfing and dark green leaves with white enations are the major symptoms of MRDD. Previous studies on the response of maize to RBSDV
infection have revealed that the expression patterns of cell wall- and development-related genes, chloroplast-related genes, and disease resistance- and stress-related genes are
dramatically altered52,53. Some genes were identified within the genomic region associated with MRDD resistance, such as cell wall-related genes GRMZM2G478892 and GRMZM2G471321, and
chloroplast-related genes GRMZM2G178602, GRMZM2G156422, GRMZM2G175867, and GRMZM2G161673. In addition to the cellular component genes, the ubiquitin-related genes and genes involved in the
ubiquitin biosynthesis pathway were influenced in maize infected with MRSDV52,53. Previous studies have also shown that protein ubiquitin-mediated degradation is involved in plant disease
resistance54,55. In the MRDD resistance-associated genomic region, two ubiquitin-related genes, namely GRMZM2G091490 and GRMZM2G110983, were predicted to be associated with the regulation of
plant growth and development and plant defense32,33. Confirmation of the roles and functions of these candidate genes is necessary to facilitate a better understanding of how these genes
contribute to MRDD resistance in maize and, more generally, improve our knowledge of the genetic mechanisms underlying resistance to viruses in higher plants. Additional studies elucidating
the mechanism of interaction between maize MRDD resistance candidate genes and RBSDV infection are necessary. Taken together, the results of the present study have provided novel insights
into the genetic architecture of MRDD resistance, which is controlled by a dominant gene in maize line K36. The susceptibility to MRDD may thus easily be controlled by identifying a
resistance gene in the host that renders resistance to the disease. Furthermore, two molecular markers, 6F29R29 and 6F34R34 on chromosome 6, were determined to be tightly linked to the
resistance gene. These two markers, together with the highly resistant inbred line K36, may potentially be utilized in MRDD resistance breeding programs. MATERIALS AND METHODS PLANT
MATERIALS The parents used in this study are elite inbred lines with high general combining ability. The maternal line S221, which is highly susceptible to MRDD, was derived from the Reid
group. The paternal line K36, which is highly resistant to MRDD, was selected from naturally mixed pollination of the US hybrid P78646 and Y7865. The F1, F2 progenies, which were selfed,
were obtained by crossing S221 with K36. Together with the two parents, these comprised the dataset employed in this study. FIELD EVALUATION The MRDD resistance of all the parental plants
and their progenies were evaluated under natural infection conditions in 2015 and 2016 in Shijiazhuang, where MRDD is prevalent. The previous crop in the experimental field was wheat, which
constitutes a reservoir for both vector and virus. Forty plants of each parent and 40 F1 individuals were planted in 2-row plots in each year. The F2 populations included 513 individual
plants in 2015 and 940 in 2016. All plants for resistance identification were sowed on May 11 of each year during the transmission period of the causal virus by its insect vector, _L_.
_striatellus_. The other two F2 populations from the same cross and their parental inbred lines were planted as controls without virus infection. One population consisting of 180 individuals
was planted on September 15, 2016 in a protected greenhouse. The other population for RIL population construction consisting of 846 individuals was sowed on June 20, 2016 to avoid
planthopper infection. All seeds were sowed by hand in rows of 0.6 m width and 5 m length. The seeds were coated with chemical agents pre-sowing to comprehensively control underground pests
and pathogenic bacteria during the germination and seedling stage. Chemical agents included bactericides of fludioxonil, metalaxyl-M, and difenoconazole, and pesticides of Phoxim and
Chlorpyrifos. The experimental field comprised cinnamon soil. Within the top-20-cm of the soil layer, the soil organic matter content was 16.2–16.9 g/kg, total nitrogen 1.14–1.20 g/kg, total
phosphorus 2.1–2.5 g/kg, alkaline hydrolysis nitrogen 128–144 mg/kg, effective phosphorus 24.40~27.84 mg/kg, and available potassium 163.5~177.4 mg/kg. The amount of base fertilizer
(composed N-P2O5-K2O (N/P/K = 22/8/10) used was 60 g/m2 without top dressing throughout the plant growing season. Irrigation and weeding during growth were the same as in the conventional
field. PHENOTYPING AND CHI-SQUARE TEST The disease severity of all tested maize plants was assessed during the maturity stage and used a four-point disease severity scheme: 0, no symptoms;
1, mild symptoms such as presence of galls on the abaxial side leaves, or enations; 2, shortened superior internodes, enations, and ‘hockey pole’ ears; and 3, severe dwarfing, enations, and
small ears with few or no kernels. Then, the disease incidence and DSI of each plot were calculated using the following equation: DSI (%) = ∑(rating scale score × number of plants with that
rating scale) × 100/3 × total number of plants as described by Di Renzo16. The F2 population was subjected to a chi-square goodness-of-fit test. VIRUS DETECTION BY RT-PCR A total of 63 plant
samples including the parental materials (K36, S221-S with MRDD symptoms, and S221 planted in a greenhouse without planthopper infection) and a subset of 60 F2 individuals making up the R-
and S-pool for deep sequencing were analyzed for the presence of the virus by RT-PCR4,56, with modifications. Approximately 100 mg of maize leaves were ground in liquid nitrogen and mixed
with 1 mL of TRIzol reagent (Invitrogen, CA, USA) for RNA extraction. The concentration and quality of RNA were estimated using a NanoDrop spectrophotometer (Thermo Scientific, Waltham, MA,
USA) and gel electrophoresis. Approximately 2 μg of total RNA was used for cDNA synthesis using a Superscript III first-strand synthesis system (Invitrogen, CA, USA) and random primers (New
England Biolabs, UK) following the manufacturer’s instructions. The virus cDNA was kindly provided by Dr. Jianfeng Wei of the Institute of Crop Science, Chinese Academy of Agricultural
Sciences. A set of published primers (up-5′AGCGGAGAACGTTTGGATC3′/dn-5′TTAACAACAGCAGCTTCACC3′) designed from the highly conserved regions (568 bp) within the viral genome was used for virus
detection4,56. RT-PCR was performed in 96 well-plates; each reaction was conducted in a 20-μL reaction containing 7.5 nM primers and 2.5 μL of cDNA. The CFX96 Connect™ Real-Time PCR
Detection System (Bio-Rad, USA) was used with the following conditions: an initial denaturation at 95 °C for 5 min, followed by 40 cycles of 94 °C for 30 s, 55 °C for 30 s, and an extension
at 72 °C for 30 s. The _Zm-Actin_ gene was used as an internal control. The primer sequences of the _Actin_ gene were up-5′CACCTTCTACAACGAGCTCC3′/dn-5′CAGTCAGGATCTTCATGAGG3′ 57. The PCR
products were assessed for purity and size by ethidium bromide staining after agarose gel electrophoresis (1.5% agarose, TAE buffer) and sequenced to confirm the presence of the target DNA
fragment. DNA EXTRACTION AND POOL CONSTRUCTION Based on disease severity, 30 extremely resistant (disease score 0) individual plants and 30 extremely susceptible (disease score 3) individual
plants from the F2 population were selected and grouped as two bulks, namely resistant pool (R-pool) and susceptible pool (S-pool), for the BSA of the year 201542. Total genomic DNA was
isolated from the young leaves of the two parents and each plant from the two bulks according to Doyle _et al_. (1990), with minor modifications58. The DNA quantity and quality were measured
with a spectrophotometer (NanoDrop 2000, Thermo Scientific, Waltham, MA USA) and by 1.5% agarose gel electrophoresis. The bulked DNA samples were prepared by mixing an equal ratio of DNA
from each plant at a final concentration of 30 ng/μL. These DNA samples were used for SLAF-seq analysis. SLAF LIBRARY CONSTRUCTION AND SEQUENCING The genome sequence of _Z_. _mays_ (2,500
Mb) was used as a reference sequence (ftp://ftp.ensemblgenomes.org/pub/plants/release-24/fasta/zea_mays/). To obtain an even distribution of SLAFs, an initial simulated restriction enzyme
digestion was conducted with the maize genomic reference sequence to optimize conditions for SLAF yield. Based on the results of the simulated restriction enzyme digestion, the genomic DNA
of the two parents and pools were digested using the appropriate restriction enzyme combination of _HaeIII_ and _Hpy166II_, with the genomic DNA of rice (_Oryza sativa_) used as a control,
to assess the effectiveness of the enzyme digestion. All subsequent SLAF-seq procedures were performed according to Sun _et al_.59 with minor modifications. DNA fragments 414–444 bp in
length were selected as SLAFs and prepared for pair-end sequencing on an Illumina High-seq. 2500 sequencing platform (Illumina, Inc.; San Diego, CA, USA) at the Beijing Biomarker
Technologies Corporation (http://www.biomarker.com.cn). SEQUENCING DATA ANALYSIS AND SLAF DEFINITION To ensure the quality and effectivity of the original sequencing data, the 100-bp
flanking regions of the raw reads of 414–444 bp in length were filtered out and used for data evaluation. SOAP2 (Short Oligo Nucleotide Alignment Program 2) was employed to map these onto
the reference genome after correction (ftp://ftp.ensemblgenomes.org/pub/plants/release-24/fasta/zea_mays/)60. The SLAF groups were generated according to the reads that were mapped to the
same position. Alleles were assigned to each SLAF based on the results of the Burrows-Wheeler Aligner (BWA) evaluation. SLAF-tags were defined as parent sequences, and bulks were genotyped
by similarity to the reference genome sequence. The GATK (Genome Analysis Toolkit) software (https://software.broadinstitute.org/gatk/best-practices?bpm=DNAseq#variant-discovery-ovw) was
used for SNP detection. First, GATK was used in conducting local realignments according to the localization results of the sequencing reads onto the reference genome, and then GATK and
Samtools (Sequence Alignment/Map tools) were used for variant SNP discovery analysis. Finally, a set of SNP sites was obtained by selecting the intersection of the SNP discovered by GATK and
Samtools to ensure accurate SNP detection. ASSOCIATION ANALYSIS The SNP-index is a recently developed association analysis method for finding significant differences in genotype frequencies
between pools indicated by Δ(SNP-index)61. In the association analysis, auj stands for the R-pool and aui for the S-pool. The Δ(SNP-index) was calculated as follows: SNP-index (auj) =
ρx/(ρX + ρx), SNP-index (aui) = ρx/(ρX + ρx), Δ(SNP-index) = SNP-index(auj) - SNP-index(aui), in which ρX and ρx represent the reads of the genotype for a single marker (SNP) in the R-pool
and S-pool, respectively. The Δ(SNP-index) was used for the association analysis. To ensure the reliability of the related association regions, ED was also used to identify significantly
different markers and evaluate the associated regions according to the method described by Hill62. Regions above the threshold were considered as trait-related candidate regions. NARROWING
DOWN THE MRDD RESISTANCE-ASSOCIATED REGIONS BY SSR ASSAYS Based on the physical locations of the SSR markers developed by Xu _et al_.63, 35 SSR markers were selected from the associated
region on chromosome 6 (Supplementary Table S4). These primers were synthesized by Sangon Biotech Co. Ltd., Shanghai, China. To obtain polymorphic SSR markers between S221 and K36, the
selected markers were first surveyed using the two parental lines. The identified informative SSR markers were then used in genotyping the F2 individuals. PCR was performed in 96-well plates
using 20-μL reactions containing 100 ng of DNA template, 1 pmol of each primer, and 2× Tag PCR StarMix with loading buffer (Invitrogen Biotechnology Co. Ltd., Shanghai, China). PCR
conditions were as follows: 1 cycle at 95 °C for 10 min, followed by 36 cycles at 95 °C for 30 s, annealing temperature at 53–58 °C for 30 s, and elongation at 72 °C for 30 s, then 1 cycle
consisting of a final elongation step at 72 °C for 10 min. All PCR products were evaluated by denaturing polyacrylamide gel electrophoresis (6% polyacrylamide) using DYY-12C (Liu-Yi,
Beijing, China) followed by silver staining. The genotype of each SSR marker was surveyed. Then, the obtained genotype data were analyzed with MAPMAKER/EXP version 3.0b64, using the Kosambi
map function to calculate genetic distances. Linkage was determined at a logarithm of the odds (LOD) threshold of 3.0, with a maximum map distance of 50 centiMorgan (cM). GO ANALYSIS OF
SELECTED CANDIDATE GENES To annotate the genes in the MRDD resistance-associated region, BLAST was used to compare these with genes in the NCBI non-redundant (NR), Swiss-Prot, GO, Clusters
of Orthologous Groups of proteins (COG), and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases, respectively. All genes that fell into three categories, including biological process,
cellular component, and molecular function, were filtered out based on the statistical information described by Harris _et al_.65. The candidate genes were selected after the analysis of the
relationship between the functions of the genes and resistance to MRDD. REFERENCES * Tao, Y., Liu, Q. & Xu, M. Research progress on maize rough dwarf disease. _J Maize Sci_ 21, 149–152
(2013). CAS Google Scholar * Shi, L.-y. _et al_. Identification of a major quantitative trait locus for resistance to maize rough dwarf virus in a Chinese maize inbred line X178 using a
linkage map based on 514 gene-derived single nucleotide polymorphisms. _Mol Breed_ 30, 615–625 (2012). Article CAS Google Scholar * Lu, H. _et al_. The effect of cause of maize rough
dwarf diease and yeild of fresh ear druing different sowing. _Jiangsu Agric. Sci._ 41, 75–76 (2014). Google Scholar * Dovas, C., Eythymiou, K. & Katis, N. First report of maize rough
dwarf virus (MRDV) on maize crops in Greece. _Plant Pathol_ 53, 238–238 (2004). Article Google Scholar * Arneodo, J., Guzmán, F., Conci, L., Laguna, I. & Truol, G. Transmission
features of Mai de Rio Cuarto virus in wheat by its planthopper vector Delphacodes kuscheli. _Ann Appl Biol_ 141, 195–200 (2002). Article Google Scholar * Bai, F. _et al_. Phylogenetic
analysis reveals that a dwarfing disease on different cereal crops in China is due to rice black streaked dwarf virus (RBSDV). _Virus Genes_ 25, 201–206 (2002). Article CAS PubMed Google
Scholar * Fang, S. _et al_. Identification of rice black-streaked dwarf fijivirus in maize with rough dwarf disease in China. _Arch Virol_ 146, 167–170 (2001). Article CAS PubMed Google
Scholar * Yin, X. _et al_. Molecular characterization of segments S7 to S10 of a southern rice black-streaked dwarf virus isolate from maize in northern China. _Virol Sin_ 26, 47–53 (2011).
Article CAS PubMed Google Scholar * Guo, Q., Li, Z. & Dong, Z. Observation and analysis of varietal resistance of maize rough dwarf virus disease (MRDV). _Plant Prot_ 1, 21–23
(1995). Google Scholar * Shang, Y. _et al_. Identification and investigation on resistance to virus diseases of both maize commercial varieties and germplasm at seedling stage. _Shandong
Agric Sci._ 4, 3–5 (2001). Google Scholar * Yongkun, C., Xinhai, L. & Muji, X. Genetic variation in sixty-four maize inbred lines in relation to maize rough dwarf virus. _Acta Agron
Sin_ (2006). * Wang, G. Y. _et al_. Identification on disease resistance of maize varieties (lines) to maize rough dwarf virus. _Acta Agric Zhejiangensis_ 3, 027 (2011). Google Scholar *
Xue, L. _et al_. Mining and analyzing genetic diversity for maize rough dwarf disease resistant gerplasms and its application in maize breeding. _Acta Agron Sin_ 37, 2123–2129 (2011).
Article ADS CAS Google Scholar * Liu, Z., Chi, S. & Qin, Z. Resistance of corn genotypes to maize rough dwarf virus. _J Maize Sci_ 4, 68–70 (1996). Google Scholar * Wang, A. _et
al_. Studies on genetic basis and recurrent selection effect of inbred line maize resistance to MRDV. _J Maize Sci_ 8, 80–82 (2000). Google Scholar * Di Renzo, M. A. _et al_. Inheritance of
resistance to Mal de Río Cuarto (MRC) disease in _Zea mays_ (L.). _J Agric Sci_ 139, 47–53 (2002). Article Google Scholar * A. Rossi, E., L. Borghi, M., A. Di Renzo, M. & C. Bonamico,
N. Quantitative Trait loci (QTL) Identification for resistance to Mal de Rio Cuarto Virus (MRCV) in maize based on segregate population. _Open Agri J_ 9 (2015). * Di Renzo, M. A. _et al_.
Microsatellite markers linked to QTL for resistance to Mal de Río Cuarto disease in _Zea mays_ L. _J Agri Sci_ 142, 289–295 (2004). Article Google Scholar * Bonamico, N. _et al_. QTL
analysis of resistance to Mal de Río Cuarto disease in maize using recombinant inbred lines. _J Agric Sci._ E150, 619–629 (2012). Article Google Scholar * Kreff, E. _et al_. Resistance to
Mal de Río Cuarto virus in maize: A QTL mapping analysis. _J Basic Appl Genet_ 17, 41–50 (2006). Google Scholar * Luan, J., Wang, F., Li, Y., Zhang, B. & Zhang, J. Mapping quantitative
trait loci conferring resistance to rice black-streaked virus in maize (_Zea mays_ L.). _Theor Appl Genet_ 125, 781–791 (2012). Article CAS PubMed Google Scholar * Tao, Y. _et al_.
Identification and fine-mapping of a QTL, _qMrdd1_, that confers recessive resistance to maize rough dwarf disease. _BMC Plant Biol_ 13, 1 (2013). Article Google Scholar * Liu, C. _et al_.
Fine mapping of a quantitative trait locus conferring resistance to maize rough dwarf disease. _Theor Aappl Genet_ 129, 2333–2342 (2016). Article CAS Google Scholar * Liu, C. _et al_.
Genetic properties of 240 maize inbred lines and identity-by-descent segments revealed by high-density SNP markers. _Mol Breed_ 35, 146 (2015). Article CAS Google Scholar * Liu, C. _et
al_. Genome-wide association study of resistance to rough dwarf disease in maize. _Eur J Plant Pathol_ 139, 205–216 (2014). Article CAS Google Scholar * Flint‐Garcia, S. A. _et al_. Maize
association population: a high-resolution platform for quantitative trait locus dissection. _Plant J_ 44, 1054–1064 (2005). Article PubMed Google Scholar * Zhu, C., Gore, M., Buckler, E.
S. & Yu, J. Status and prospects of association mapping in plants. _Plant Genome_ 1, 5–20 (2008). Article CAS Google Scholar * Korte, A. & Farlow, A. The advantages and
limitations of trait analysis with GWAS: a review. _Plant Methods_ 9, 29 (2013). Article CAS PubMed PubMed Central Google Scholar * Chen, G., Wang, X., Hao, J., Yan, J. & Ding, J.
Genome-wide association implicates candidate genes conferring resistance to maize rough dwarf disease in maize. _PloS One_ 10, e0142001 (2015). Article PubMed PubMed Central Google
Scholar * Hao, D. _et al_. Identification of significant single nucleotide polymorphisms for resistance to maize rough dwarf disease in elite maize (_Zea mays_ L.) inbred lines. _Euphytica_
203, 109–120 (2015). Article ADS CAS Google Scholar * Cleary, A. L. & Smith, L. G. The Tangled1 gene is required for spatial control of cytoskeletal arrays associated with cell
division during maize leaf development. _Plant Cell_ 10, 1875–1888 (1998). Article CAS PubMed PubMed Central Google Scholar * Liu, Z., Shao, F., Tang, G., Shan, L. & Bi, Y. Cloning
and characterization of a transcription factor _ZmNAC1_ in maize (_Zea mays_). _Yi Chuan = Hereditas_ 31, 199–205 (2009). Article CAS PubMed Google Scholar * Mao, H., Yu, L., Han, R.,
Li, Z. & Liu, H. ZmNAC55, a maize stress-responsive NAC transcription factor, confers drought resistance in transgenic. _Arabidopsis. Plant Physiol Bioch_ 105, 55–66 (2016). Article CAS
Google Scholar * Marchler-Bauer, A. _et al_. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. _Nucleic Acids Res_ 45, D200–D203 (2016). Article
PubMed PubMed Central Google Scholar * Thomas, B. R., Romero, G. O., Nevins, D. J. & Rodriguez, R. L. New perspectives on the endo-beta-glucanases of glycosyl hydrolase Family 17.
_Int J Biol Macromol_ 27, 139–144 (2000). Article CAS PubMed Google Scholar * Nawaz, G. & Kang, H. Chloroplast-or Mitochondria-Targeted DEAD-Box RNA Helicases Play Essential Roles in
Organellar RNA Metabolism and Abiotic Stress Responses. _Front Plant Sci_ 8 (2017). * Fraering, P. _et al_. The GPI transamidase complex of Saccharomyces cerevisiae contains Gaa1p, Gpi8p,
and Gpi16p. _Mol Biol Cell_ 12, 3295–3306 (2001). Article CAS PubMed PubMed Central Google Scholar * Castellano, M. M. & Sablowski, R. Phosducin-like protein 3 is required for
microtubule-dependent steps of cell division but not for meristem growth in _Arabidopsis_. _Plant Cell_ 20, 969–981 (2008). Article CAS PubMed PubMed Central Google Scholar * Myles, S.
_et al_. Association mapping: critical considerations shift from genotyping to experimental design. _Plant Cell_ 21, 2194–2202 (2009). Article CAS PubMed PubMed Central Google Scholar *
Xu, Q. _et al_. Whole-genome expression analysis of Rice black-streaked dwarf virus in different plant hosts and small brown planthopper. _Gene_ 572, 169–174 (2015). Article CAS PubMed
Google Scholar * Wing, R. A., Zhang, H.-B. & Tanksley, S. D. Map-based cloning in crop plants. Tomato as a model system: I. Genetic and physical mapping of jointless. _Mol Gen Genet_
242, 681–688 (1994). Article CAS PubMed Google Scholar * Michelmore, R. W., Paran, I. & Kesseli, R. Identification of markers linked to disease-resistance genes by bulked segregant
analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. _P Natl Acad Sci USA_ 88, 9828–9832 (1991). Article ADS CAS Google Scholar *
Takagi, H. _et al_. QTL‐seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. _Plant J_ 74, 174–183 (2013). Article CAS
PubMed Google Scholar * Trick, M. _et al_. Combining SNP discovery from next-generation sequencing data with bulked segregant analysis (BSA) to fine-map genes in polyploid wheat. _BMC
Plant Biol_ 12, 14 (2012). Article CAS PubMed PubMed Central Google Scholar * Yang, Z. _et al_. Mapping of quantitative trait loci underlying cold tolerance in rice seedlings via
high-throughput sequencing of pooled extremes. _PloS One_ 8, e68433 (2013). Article ADS CAS PubMed PubMed Central Google Scholar * Han, Y. _et al_. Combining next generation sequencing
with bulked segregant analysis to fine map a stem moisture locus in sorghum (_Sorghum bicolor_ L. Moench). _PloS One_ 10, e0127065 (2015). Article PubMed PubMed Central Google Scholar *
Livaja, M. _et al_. BSTA: a targeted approach combines bulked segregant analysis with next-generation sequencing and de novo transcriptome assembly for SNP discovery in sunflower. _BMC
Genomics_ 14, 628 (2013). Article CAS PubMed PubMed Central Google Scholar * Yu, P. _et al_. A maize bundle sheath defective mutation mapped on chromosome 1 between SSR markers umc1395
and umc1603. _J Integr Agr_ 14, 1949–1957 (2015). Article Google Scholar * Xia, C. _et al_. Identification of a new maize inflorescence meristem mutant and association analysis using
SLAF-seq method. _Euphytica_ 202, 35–44 (2015). Article Google Scholar * Bonamico, N. C. _et al_. Association between microsatellites and resistance to Mal de Río Cuarto in maize by
discriminant analysis. _Phyton-Revista Int de Bot Experimental_ 79, 31 (2010). Google Scholar * Wang, F. & Zhang, Y. Molecular mapping of three loci conferring resistance to maize rough
dwarf disease. _Mol Plant Breed_ 5, 178–179 (2007). CAS Google Scholar * JIA, M. A. _et al_. Alteration of gene expression profile in maize infected with a double‐stranded RNA fijivirus
associated with symptom development. _Mol Plant Pathol_ 13, 251–262 (2012). Article CAS PubMed Google Scholar * Zhou, Y. _et al_. Dual transcriptome analysis reveals insights into the
response to Rice black-streaked dwarf virus in maize. _J Exp Bot_ 67, 4593–4609 (2016). Article CAS PubMed PubMed Central Google Scholar * Abreu, P. _et al_. A current overview of the
Papaya meleira virus, an unusual plant virus. _Viruses_ 7, 1853–1870 (2015). Article CAS PubMed PubMed Central Google Scholar * Soosaar, J. L., Burch-Smith, T. M. & Dinesh-Kumar, S.
P. Mechanisms of plant resistance to viruses. _Nat Rev Microbiol_ 3, 789 (2005). Article CAS PubMed Google Scholar * Ortiz, V., Lucas, J. B., QUEROL, A. L. & Romero, J. Detection of
maize rough dwarf virus in Spain: a survey of susceptible host genotypes and molecular characterization of two genomic segments of the virus. _Phytopathol Mediterr_ 53, 40–53 (2014). CAS
Google Scholar * Yue, R. _et al_. Genome-wide identification and expression profiling analysis of _ZmPIN_, ZmPILS, ZmLAX and ZmABCB auxin transporter gene families in maize (_Zea mays_ L.)
under various abiotic stresses. _Plos One_ 10, e0118751 (2015). Article PubMed PubMed Central Google Scholar * Doyle, J. & Doyle, J. Isolation of DNA from small amounts of plant
tissues. _BRL focus_ 12, V15 (1990). Google Scholar * Sun, X. _et al_. SLAF-seq: an efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing.
_PloS One_ 8, e58700 (2013). Article ADS CAS PubMed PubMed Central Google Scholar * Li, R. _et al_. SOAP2: an improved ultrafast tool for short read alignment. _Bioinformatics_ 25,
1966–1967 (2009). Article CAS PubMed Google Scholar * Fekih, R. _et al_. MutMap + : genetic mapping and mutant identification without crossing in rice. _PloS One_ 8, e68529 (2013).
Article ADS CAS PubMed PubMed Central Google Scholar * Hill, J. T. _et al_. MMAPPR: mutation mapping analysis pipeline for pooled RNA-seq. _Genome Res_ 23, 687–697 (2013). Article CAS
PubMed PubMed Central Google Scholar * Xu, J. _et al_. Development and characterization of simple sequence repeat markers providing genome-wide coverage and high resolution in maize.
_DNA Res_ 20, 497–509 (2013). Article CAS PubMed PubMed Central Google Scholar * Lincoln, S. E., Daly, M. J. & Lander, E. S. Constructing genetic linkage maps with MAPMAKER/EXP
Version 3.0: a tutorial and reference manual. _A Whitehead Institute for Biomedical Research Technical Report_, 78–79 (1993). * Harris, M. _et al_. The Gene Ontology (GO) database and
informatics resource. _Nucleic Acids Res_ 32, D258–D261 (2004). Article ADS CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS The Doctoral Fund of the Hebei Academy of
Agriculture and Forestry Sciences (No. BS201504); Program of Agricultural Modern Technology of Hebei Province (F17C10007), and The National Key Research and Development Program of China
(2016YFD0101205–6) supported this study. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Key Laboratory of Crop Genetics and Breeding of Hebei Province, Institute of Cereal and Oil Crops,
Hebei Academy of Agriculture and Forestry Sciences, Shijiazhuang, 050035, China Ronggai Li, Wei Song, Baoqiang Wang, Jianghao Wang, Dongmin Zhang, Quanguo Zhang, Xinghua Li, Jianfen Wei
& Zengyu Gao Authors * Ronggai Li View author publications You can also search for this author inPubMed Google Scholar * Wei Song View author publications You can also search for this
author inPubMed Google Scholar * Baoqiang Wang View author publications You can also search for this author inPubMed Google Scholar * Jianghao Wang View author publications You can also
search for this author inPubMed Google Scholar * Dongmin Zhang View author publications You can also search for this author inPubMed Google Scholar * Quanguo Zhang View author publications
You can also search for this author inPubMed Google Scholar * Xinghua Li View author publications You can also search for this author inPubMed Google Scholar * Jianfen Wei View author
publications You can also search for this author inPubMed Google Scholar * Zengyu Gao View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS
Conceived and designed the experiments: R.L., W.S. and B.W. Performed the experiments: R.L., J.W., D.Z., Q.Z, X.L., J.W. and Z.G. Analyzed the data: R.L. and J.W. Contributed materials: W.S.
and B.W. Wrote the manuscript: R.L. Reference collection and data management: R.L., J.W. and Q.Z. CORRESPONDING AUTHOR Correspondence to Ronggai Li. ETHICS DECLARATIONS COMPETING INTERESTS
The authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER'S NOTE: Springer Nature remains neutral with regard to jurisdictional claims in published maps and
institutional affiliations. ELECTRONIC SUPPLEMENTARY MATERIAL SUPPLEMENTARY INFORMATION RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0
International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the
source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Li, R., Song, W., Wang, B. _et al._ Identification of a locus conferring dominant
resistance to maize rough dwarf disease in maize. _Sci Rep_ 8, 3248 (2018). https://doi.org/10.1038/s41598-018-21677-3 Download citation * Received: 17 August 2017 * Accepted: 07 February
2018 * Published: 19 February 2018 * DOI: https://doi.org/10.1038/s41598-018-21677-3 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get
shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative