Rudhira/bcas3 is essential for mouse development and cardiovascular patterning
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Rudhira/Breast Carcinoma Amplified Sequence 3 (BCAS3) is a cytoskeletal protein that promotes directional cell migration and angiogenesis _in vitro_ and is implicated in human
carcinomas and coronary artery disease. To study the role of Rudhira during development _in vivo_, we generated the first knockout mouse for _rudhira_ and show that Rudhira is essential for
mouse development. _Rudhira_ null embryos die at embryonic day (E) 9.5 accompanied by severe vascular patterning defects in embryonic and extra-embryonic tissues. To identify the molecular
processes downstream of _rudhira_, we analyzed the transcriptome of intact knockout yolk sacs. Genome-wide transcriptome analysis showed that Rudhira functions in angiogenesis and its
related processes such as cell adhesion, extracellular matrix organization, peptidase activity and TGFβ signaling. Since Rudhira is also expressed in endothelial cells (ECs), we further
generated Tie2Cre-mediated endothelial knockout (CKO) of _rudhira_. CKO embryos survive to E11.5 and similar to the global knockout, display gross vascular patterning defects, showing that
endothelial Rudhira is vital for development. Further, Rudhira knockdown ECs in culture fail to sprout in a spheroid-sprouting assay, strongly supporting its role in vascular patterning. Our
study identifies an essential role for Rudhira in blood vessel remodeling and provides a mouse model for cardiovascular development. SIMILAR CONTENT BEING VIEWED BY OTHERS ETV2 FUNCTIONS AS
A PIONEER FACTOR TO REGULATE AND REPROGRAM THE ENDOTHELIAL LINEAGE Article 12 May 2022 MED23 SUPPORTS ANGIOGENESIS AND MAINTAINS VASCULAR INTEGRITY THROUGH NEGATIVE REGULATION OF
ANGIOPOIETIN2 EXPRESSION Article Open access 19 April 2022 EPIGENETIC LANDSCAPE REVEALS MECOM AS AN ENDOTHELIAL LINEAGE REGULATOR Article Open access 25 April 2023 INTRODUCTION The
cytoskeleton is composed of microtubules, microfilaments and intermediate filaments that regulate each other and are intimately linked in several normal and diseased contexts1. Cytoskeletal
reorganization is a major requirement in processes that require change in cell adhesion and migration status2,3,4. This is accompanied by change in cell shape and gene expression through a
complex network of signaling pathways by influencing transcription factor activity or otherwise. Homozygous mutants of tubulins and microtubule-associated proteins (MAPs) show
pre-gastrulation lethality on the one hand or display mild developmental phenotypes on the other5,6,7. However, the functions of a large number of cytoskeletal proteins remain to be
elucidated. Rudhira is a cytoskeletal WD40 domain containing protein that binds microtubules and promotes directional cell migration _in vitro_ by activating Cdc42 for actin reorganization
and filopodial extension8. Rudhira knockdown leads to random and retarded cell migration _in vitro_ whereas overexpression promotes migration in non-motile cells. Rudhira is expressed in
mouse embryonic stem cells, primitive erythropoiesis and vascular precursors, endothelial cells (ECs), in human vascular development in an _in vitro_ embryoid body model and in malignant
tumors and blood vessels8,9,10. Human Rudhira/Breast Cancer Amplified Sequence 3 (BCAS3) has recently been associated with coronary artery disease11. To elucidate the normal role of Rudhira
_in vivo_ we generated _rudhira_ mutant mice using Cre-loxP mediated deletion and analyzed the consequence of _rudhira_ deficiency _in vivo_. We report for the first time that _rudhira_
deletion results in mid-gestation lethality with aberrant cardiovascular patterning. Rudhira deletion causes aberrant gene expression as seen by yolk sac transcriptome studies. We show that
endothelial Rudhira is essential for angiogenesis and vascular remodeling during development. RESULTS _RUDHIRA_ IS VITAL FOR EMBRYONIC DEVELOPMENT Rudhira is expressed primarily in early
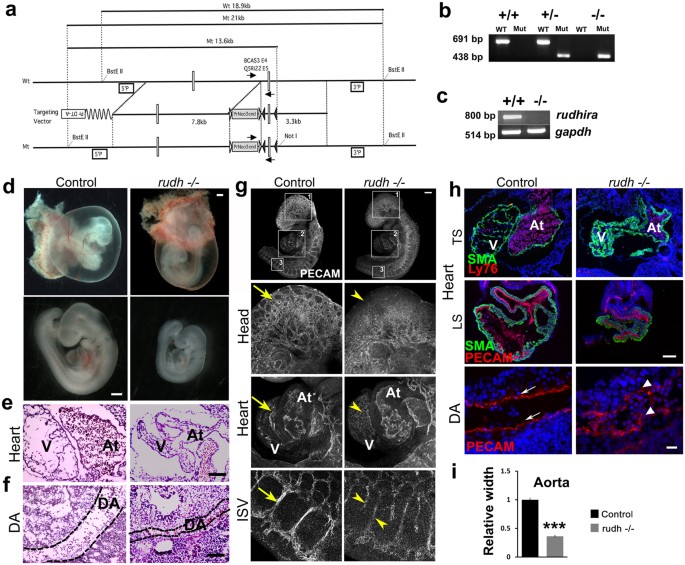
embryonic vascular development and neo-angiogenesis, but its role _in vivo_ is not known. Hence we generated _rudhira_ floxed mice (Fig. 1a and Fig. S1a) and crossed them to _CMV-Cre_ for
ubiquitous deletion (_rudhira__flox/flox_; _CMVCre_+ abbreviated to _rudh__−/−_) (see Materials and methods and Fig. S1a). While heterozygotes were viable, ubiquitous deletion of _rudhira_
gave no live homozygous pups (Table S1a). This indicates that deletion of _rudhira_ causes recessive embryonic lethality. Analysis of embryos from E8.5 to E11.5 showed a reduced number of
homozygous mutant embryos (Table S1a) as identified by genotyping (Fig. 1b), transcript (Fig. 1c) and protein (Fig. S1b,c) expression, suggesting that lethality occurred between E9.0 and
E10. Chi-square test showed significantly reduced frequency of knockout embryos from E8.5 onwards, as compared to the expected. Since _rudhira_ expression may be transient or undetectable in
some migrating cells we also analyzed the effect of globally deleted _rudhira_ (_rudh_−/−) on development from E7.5 onwards, a stage before _rudhira_ expression is detectable10. At E7.5
_rudhira_ mutant embryos were indistinguishable from littermate control with respect to morphology as well as primitive streak formation as seen by Brachyury expression (Fig. S1d). However,
at E8.5, the mutant embryos showed unpatterned dorsal aorta as detected by Flk1 staining (Fig. S1e-e’). By E9.5, mutant embryos were often growth retarded (20/51 = 39.2%) (Fig. 1d and Fig.
S1f) with defects including reduced somite number (19 ± 2 at E9.5 in growth retarded mutants compared to 25 ± 2 in controls; n = 10). This suggests that Rudhira may be essential for multiple
developmental processes and hence its depletion leads to embryonic lethality. RUDHIRA PLAYS A KEY ROLE IN CARDIOVASCULAR DEVELOPMENT AND TISSUE PATTERNING In mouse development Rudhira is
known to have restricted expression during vasculogenesis and primitive erythropoiesis10. Hence, we reasoned that cardiovascular defects could be one major cause of growth retardation and
lethality seen in _rudhira_ null embryos. To investigate this further, we analyzed the effect of _rudhira_ deletion on cardiac and vascular patterning (Fig. 1e–i). Whole mount immunostaining
of _rudh_−/− embryos with anti-PECAM1 antibodies showed striking defects in the morphology and vasculature of the head and heart in all mutant embryos analyzed at E9.5 even if they were not
developmentally delayed (Fig. 1g). While control embryos had a well formed vascular network comprising major vessels giving rise to intricate secondary and tertiary branches (Fig. 1g,
arrows), _rudhira_ mutants showed completely disorganized head vasculature with defective vessel sprouting, reduced capillaries and impaired branching of intersomitic vessels (ISVs) that
failed to sprout into fine capillaries (Fig. 1g arrowheads). Histological analysis (Fig. 1e,f) as well as immunostaining for cardiovascular markers (Fig. 1h) showed that _rudh__−/−_ embryos
had collapsed, smaller heart chambers, reduced endocardium development and a fused atrio-ventricular canal. Dorsal aorta was discontinuous with a pronounced decrease in the lumen and
intersomitic vessels were improperly patterned (Fig. 1f–i). The endothelial lining was disorganized in all tissues and ECs seemed to have impaired or random migration and were unable to form
organized vessels (Fig. 1g,h). These observations show that Rudhira is essential for development and its loss leads to defects in cardiac and vascular patterning. RUDHIRA ALSO FUNCTIONS IN
EXTRAEMBRYONIC VASCULAR DEVELOPMENT Impaired development and embryonic lethality between E8.5-E11.5 is often the result of aberrant and functionally impaired extra-embryonic vasculature12.
Moreover, Rudhira is strongly expressed in the yolk sac vasculature10. Hence we analyzed extraembryonic structures of mutant embryos, such as yolk sac and placenta, which connect the
maternal and fetal vasculature. Mutant yolk sacs were pale and had few major blood vessels (Fig. 1d). Immunostaining for the blood vessel marker PECAM showed that _rudh__−/−_ yolk sac
vessels were irregular and fused, unlike the finely patterned honey-comb like vascular network seen in control littermates (Figs 2a, S2). Thus mutants could form a primitive vascular plexus
which, however, did not undergo angiogenic remodeling. Histological analyses of yolk sac showed congested capillaries lined by thinner endothelium (Fig. 2b, arrowhead) as compared to
controls (Fig. 2b, arrow). These results indicate that _rudhira_ is essential for remodeling the yolk sac vascular network. So we reasoned that aberrant vascular remodeling in _rudhira_
mutants is likely the primary cause of death. Placental circulation is vital for nourishment and development of the embryo. Improper development of the labyrinth, the feto-maternal
interface, results in poor fetal invasion and causes growth retardation13. Morphological analyses showed that _rudhira_ null embryos have a smaller placenta with abnormal histology as
compared to controls (Fig. 2c,d). Control placenta showed a distinct chorionic plate, labyrinth, spongiotrophoblast and decidual layers. _Rudh__−/−_ placenta lacked stratified layers with a
greatly reduced labyrinth and chorionic plate composed mostly of trophoblast giant cells. Fetal blood vessels could not invade into the placenta of _rudh__−/−_ and contained fewer Ly76+
fetal erythrocytes as compared to controls where maternal (arrows) and fetal (arrowheads) blood cell pockets co-existed (Fig. 2c,d). Taken together, these findings suggest that Rudhira is
essential for fetal vessel invasion into the developing placenta. Further, growth retardation in _rudh__−/−_ embryos is likely a result of defective placental circulation. RUDHIRA CONTROLS
THE ANGIOGENESIS GENE REGULATORY NETWORK To identify the processes affected by depletion of Rudhira leading to its cardiovascular phenotypes, we performed whole transcriptome-based analysis
of gene expression in _rudhira_ knockout yolk sac and embryos at E9.5 (Fig. 3). Since Rudhira localization is dynamic and like other cytoskeletal elements, would likely change with tissue
manipulation such as isolation and culture of cells, we chose to analyze intact tissue to understand how Rudhira affects the transcriptome. While embryos at E9.5 have a diverse set of
derivatives of all three germ layers, yolk sacs are primarily made of primitive endoderm and mesoderm, the latter comprising mainly endothelial and hematopoietic lineages. Hence, we
separately analyzed _rudh__−/−_ embryos and yolk sacs. Since the role of Rudhira in endothelial cell migration and vessel formation _in vitro_ is well established8 and also because knockout
embryos have vessel patterning defects, we extensively validated the transcriptome data by quantitative PCR-based expression analysis of yolk sac and endothelial cell line (Fig. 4). Volcano
plot based method was used to visualize the transcripts that are two-fold differentially expressed in yolk sac (Fig. 3a). 3291 unique probes showed 2-fold or greater statistically
significant changes in gene expression (Table S2). Of these 546 were downregulated and 334 upregulated in embryo and 566 downregulated and 1960 upregulated in yolk sac (Fig. 3b). 29
downregulated and 43 upregulated genes were common between embryo and yolk sac (Fig. 3b and Table S3). Unsupervised hierarchical cluster analysis showed that genes with similar expression
patterns were clustered together with branch distance proportional to their similarity in expression pattern. A distinct subset showed reciprocal expression between embryo and yolk sac (Fig.
3c). Interestingly the majority of clustered genes were mainly upregulated in the yolk sac (Fig. 3c,d), while the embryo had a more balanced distribution in each cluster (Fig. 3c). To
define how changes in gene expression caused by _rudhira_ depletion may influence vascular development and remodeling, we functionally annotated the data using DAVID (Database for
Annotation, Visualization and Integrated Discovery) and found that genes linked to many biological pathways were enriched. Key deregulated biological categories were identified (Fig. 3e,
Table S4). Analysis of the entire data set showed greater variation between duplicates of embryo than yolk sac, possibly because of higher heterogeneity in the embryonic tissue. Hence for
further analysis we focused on yolk sac data as it is also one of the primary sites of vascular remodeling and shows early Rudhira expression. Significant expression changes were seen in
genes that relate to a range of processes or pathways, which could impact on vascular development and remodeling (Figs 3e, 4 and Table S5). Gene ontology analysis of the common genes
identified principal biological processes affected by the loss of _rudhira_ with a Z score above 2.5. Important regulators of cellular processes such as angiogenesis/blood vessel remodeling,
extracellular matrix, regulation of proteolysis, negative regulation of peptidase activity and cell projection organization were identified. Further, key molecular families involved in
cytoskeletal remodeling, cell adhesion, cell migration and TGFβ and VEGF pathways, which are all important during angiogenesis, were connected by Rudhira/BCAS3 allowing us to identify the
Rudhira network in angiogenesis (Fig. 3f and Table S6). IDENTIFICATION OF REGULATORY NETWORKS AND NODES REGULATED BY RUDHIRA A total of 140 genes from cluster analysis were enriched in the
key gene ontology (GO) and pathways identified (Table S5) with a significance criterion of p < 0.05. Further, we were able to associate GOs and pathways known to co-operate during
vascular development and remodeling namely adhesion, angiogenesis, cytoskeleton, ECM organization, peptidase activity and TGFβ signaling (Fig. 3e). Genes differentially expressed in these
six processes were subjected to unsupervised hierarchical clustering to identify molecular signatures (Fig. S3a–f). An interaction network of significant GO terms was assembled into a GO map
to depict the relationship among prominent functional categories (Fig. S3a–f). Subsequent verification of expression data was carried out for key genes known to mediate these processes
(Fig. 4a–j) by transcript (Fig. 4a–h) or protein (Fig. 4i–j) analysis. 51 out of the 3407 genes that showed significant variation from control in the knockout yolk sac were validated by
qRT-PCR on cDNA generated from _rudhira_ knockdown and non-silencing control endothelial cell line RNA (Fig. 4i) and 70% of these (36/51) agreed with the high-throughput array data (Fig.
S3a–f and Fig. 4a–g). Changes in expression level of selected candidates were further validated using cDNA generated from fresh E9.5 yolk sac RNA (Fig. 4h). The protein levels of
representative molecules important for angiogenesis (Sox9, Wnt3a), adhesion (Itga4, Itga6), ECM (Fibronectin, Laminin and Collagen, MMP2, MMP9) and cytoskeletal organization (Vimentin) were
tested by western blotting (Fig. 4j and Fig. S5). We find that in all cases the protein expression data corroborates that seen by microarray or RT-PCR and is in agreement with the phenotype
observed. However, as the net effect on function cannot be assessed from a few protein levels, we also carried out functional assays on _rudhira_ depleted cells. ENDOTHELIAL DELETION OF
_RUDHIRA_ LEADS TO CARDIOVASCULAR AND EXTRAEMBRYONIC VASCULATURE DEFECTS Since Rudhira is expressed primarily in early embryonic vascular development and neo-angiogenesis and _rudhira_ null
embryos have vascular defects, we crossed _rudhira_ floxed mice with _Tie-2-Cre_ for tissue-specific ablation (_rudhira__flox/flox_; _TekCre_+ abbreviated to _rudh_CKO) of the _rudhira_
locus. _Tie2Cre/TekCre_ has been used to delete genes in endothelial and hematopoietic lineages. Since Rudhira is primarily expressed in endothelial cells, we utilized _TekCre_ to study the
function of endothelial Rudhira. However, the contribution of Rudhira expressed in the hematopoietic compartment cannot be ignored. Similar to ubiquitous knockout, _TekCre_-mediated deletion
of _rudhira_ was validated at RNA and protein levels, and gave no live homozygous pups (Table S1b, Fig. S4a–c). Dramatic reduction in the RNA and protein levels of Rudhira upon _TekCre_
mediated knockout confirmed that at this stage Rudhira is primarily expressed in the endothelial cells. Histological analysis as well as immunostaining for cardiovascular markers showed that
like _rudh__−/−_, _rudh__CKO_ embryos had collapsed, smaller heart chambers and reduced endocardium development. Also, the major blood vessels including dorsal aorta and intersomitic
vessels were discontinuous and improperly patterned (Fig. 5a–d). Whole mount immunostaining of _rudh_CKO embryos with anti-PECAM1 antibodies showed striking defects in the morphology and
vasculature of the head and intersomitic vessels (ISVs) in all mutant embryos analyzed at E10.5 (Fig. 5e). Control embryos had a well formed vascular network comprising major vessels giving
rise to intricate secondary and tertiary branches (Fig. 5e, arrows). Although less severe, _rudh_CKO embryos showed disorganized head vasculature with defective vessel sprouting, reduced
capillaries and impaired branching of intersomitic vessels (ISVs) that failed to sprout into fine capillaries (Fig. 5e arrowheads). _Rudh_CKO yolk sacs had reduced branching from major
vessels, vessel fusion and loss of branch hierarchy at both E10.5 and E11.5 (Fig. 6a). Further, placental thickness was reduced at both E10.5 and E11.5 (Fig. 6b–e). Fetal vessel invasion was
comparable to control at E10.5 (Fig. 6b,c) but reduced at E11.5 (Fig. 6d,e) although not as severely as in the global knock-out. This could explain the absence of significant growth
retardation in conditional mutant embryos. Thus _TekCre_-mediated deletion of _rudhira_ resulted in similar phenotypes as in the global knock-out. These results indicate that endothelial
_rudhira_ is essential for remodeling the vascular network. RUDHIRA IS ESSENTIAL FOR ENDOTHELIAL SPROUTING ANGIOGENESIS The embryonic lethality and reduced vascular patterning in _rudhira_
null embryos could be due to a ubiquitous role for Rudhira and/or a specific requirement in endothelial cells. In both null and CKO mutants ECs are specified but vessel patterning is
aberrant, indicating the process depends on endothelial Rudhira. Vascular pattern and remodeling requires proper endothelial cell migration and vessel sprouting. We showed earlier that
Rudhira is essential for EC migration8. As expected and in concordance to the previous cell line data, the cells derived from _rudhira_ knockout yolk sacs also showed retarded migration
rates and a loss of directionality (Fig. 7a). Transcriptome analysis revealed that Rudhira also affects adhesion and extracellular matrix remodeling, which are key processes in sprouting
angiogenesis. Angiogenesis requires localized remodeling of extracellular matrix (ECM) and cell-cell and cell-matrix adhesions, leading to cell invasion into the ECM and extension of the
vessel sprouts. In a Matrigel invasion assay _rudhira_ KD ECs showed reduced invasion (Fig. 7b). Also cell adhesion was reduced on gelatin and collagen matrices upon _rudhira_ depletion
(Fig. 7c). Finally, we tested _rudhira_ knockdown ECs in an _in vitro_ spheroid sprouting angiogenesis assay. While control cells expressing non-silencing shRNA (NS) showed primary,
secondary and tertiary sprouts that increased in length over the time course of the assay, _rudhira_ KD ECs failed to sprout (Fig. 7d). Thus Rudhira is essential for sprouting angiogenesis,
indicating that the vascular defects in the mouse knockout are primarily due to lack of endothelial Rudhira and not mere placental insufficiency or general developmental defects. DISCUSSION
Vertebrate blood vessel formation involves _de novo_ differentiation of endothelial cells (ECs) to form a primary plexus, which is pruned and patterned into a hierarchical network by
angiogenesis14. A fine balance of pro- and anti-angiogenic cues maintains ECs in a quiescent state15,16. Perturbation of this balance leads to endothelial activation and cytoskeletal changes
resulting in sprouting, migration and maturation17,18. EC migration and tube formation are key steps during angiogenesis, however, molecular mechanisms that operate are incompletely
defined. Random and retarded EC migration results in an unpatterned and often leaky vasculature19,20. We report here that global or endothelial deletion of a cytoskeletal protein, Rudhira,
resulted in mid-gestation lethality with severe defects in cardiovascular patterning. Based on a transcriptome analysis we identified key steps in blood vessel remodeling such as cell
adhesion, migration, extracellular matrix components and TGFβ signaling that are regulated by the action of Rudhira. We describe, for the first time, a regulatory network mediated by Rudhira
with reference to its interacting partners at both binary (regulatory) as well as physical levels (Fig. 3f). From E8.5 in the mouse yolk sac, blood flow dictates vessel fusion and
directional cell migration resulting in vascular remodeling21. Cardiac remodeling defects seen in _rudhira_ mutants may also impair circulation and contribute to the vascular remodeling
abnormalities. Hence all abnormalities noted in _rudhira_ null embryos could be due to placental insufficiency and generalized growth retardation. However, our analysis of the CKO mutants
shows conclusively that there is an essential role for endothelial Rudhira in vascular patterning as well as survival. Loss of Rudhira affects expression of the endothelial cell
transcriptome. Genes implicated in multiple processes important for angiogenesis such as cell adhesion, invasion, matrix organization and degradation are deregulated in the _rudhira_
knockout. It is likely that imbalance in action of these processes and aberrant signaling result in grossly defective angiogenesis in _rudhira_ knockout. Further, the TGFβ pathway, which
functions in a multitude of angiogenic processes was over-represented in the _rudhira_ knockout, suggesting a role in TGFβ signalling, though other signaling pathways cannot be ruled out.
Our analysis provides tools to investigate further the precise roles of Rudhira in the TGFβ pathway and in sprouting angiogenesis and vascular remodeling. Given the large number of signals
and their varying levels that cells encounter, it is unlikely that they respond only to one or the same concentration all across the organism. A robust response over a range of signals is
likely mediated by molecules that can crosstalk with a wide variety of processes. The cytoskeleton is ideally positioned for this role. Endothelial cells respond to a variety of signals
resulting in a limited repertoire of cytoskeletal changes, which in turn determine cell phenotype. The presence or absence of cell type-specific components such as Rudhira could provide
decisive control of endothelial cell behavior in response to a varying milieu of signals. For example, Rudhira could have distinct context-dependent tissue-specific roles in regulating the
cytoskeleton in EC migration as reported earlier8 or the transcriptome in development as reported here. As both angiogenic remodeling and sprouting angiogenesis occur in embryogenesis, our
study opens up new avenues for understanding vascular patterning in development and disease. MATERIALS AND METHODS GENERATION AND VALIDATION OF _RUDHIRA_ KNOCKOUT MICE The _rudhira_ locus
(Chromosome location: 11:85166669-85639560:1) was targeted at exon 6 by homologous recombination using a targeting vector (Fig. 1a) in TT2 mouse embryonic stem cells in order to generate a
floxed _rudhira_ allele22. Recombinants were selected by G418 resistance and genomic analyses with Southern hybridization, and microinjected into ICR (CD-1) 8-cell stage embryos. Resulting
chimeras were then crossed to C57BL/6N to obtain germ line transmitted recombinant progeny. The heterozygous recombinant alleles (_rudh__flox/_+) were then confirmed by Southern blotting
(Supplementary Fig. 1a). Matings between heterozygous _rudh__flox/+_ mice were set up to obtain homozygous floxed _rudhira_ mice. Deletion of _rudhira_ ubiquitously using _CMV-Cre_23 or
tissue- specifically by crossing to the endothelial specific _Tie-2-Cre_24 gave heterozygous knockout mice which were phenotypically normal with viability and fecundity similar to control
littermates. Heterozygotes (_rudh__fl/+_; _Cre__+_) were crossed to _rudhira_ floxed mice (_rudh__fl/fl_) and the progeny analyzed as described in Results. All animal experimental protocols
were approved by Institutional Animal Ethics Committee (IAEC) of JNCASR (Project number MSI006) and the Institutional Animal Ethics Committee (IAEC) of NCBS (Project number KVR-2(2)/2015).
All animals were maintained and experiments performed according to the guidelines of the animal ethics committees of JNCASR, NCBS and RIKEN, CDB. _Rudhira_ floxed mice (Accession No.
CDB0664K: http://www2.clst.riken.jp/arg/mutant%20mice%20list.html) and knockout mice were validated by genotyping (see Fig. 1 and Fig. S1) (For genotyping, see the supplementary Materials
and methods.). GENERATION OF KNOCKDOWN EC LINES _Rudhira_/_BCAS3_shRNA vectors (715, 716), and scrambled (non- silencing) control vector (TR30015) (Origene, USA) were microporated into SVEC
(mouse endothelial cell line) and selected for stable line generation. (For further details, see the supplementary Materials and methods.) Control cell line is referred to as ‘NS’ and
_rudhira_ knockdown as ‘KD’. QUANTITATIVE RT-PCR (QRT-PCR) Gene expression changes were quantitated by real-time PCR on cDNA generated from control or mutant RNA. (For further details, see
the supplementary Materials and methods.). Primers used are provided in Supplementary Table S7. IMMUNOSTAINING AND IMMUNOHISTOCHEMISTRY Yolk sac or embryos dissected at desired stages
between E7.5 to E11.5, were fixed in 4% paraformaldehyde and processed for cryosectioning (embryos) and immunostaining using standard procedures25. Samples were viewed and imaged using
bright field, phase contrast or fluorescence microscopy (For further details, see the supplementary Materials and methods.) WESTERN BLOT ANALYSIS 25 μg lysate from control or _rudhira_
knockdown cell lines of SVEC was used for western blot analysis by standard protocols. Blots were cut into strips and incubated with primary antibodies as indicated: MMP2, MMP9 (Cell
Signaling Technology), Fibronectin, Vimentin, GAPDH (Sigma Chemical Co., USA), Sox 9 (RND Systems), Laminin, ColIV, Wnt3a (Abcam), Itga6, Itga4 (BD Biosciences) and BCAS3 (Bethyl Labs, USA).
HRP conjugated secondary antibodies against appropriate species were used and signal developed by using Clarity Western ECL substrate (Biorad, USA). CELL MIGRATION ASSAY E9.5 yolk sacs were
washed in phosphate buffered saline (PBS), minced and dissociated in 0.2% collagenase type IV (GIBCO/BRL) at 37 °C for 5 minutes, washed, pelleted and resuspended in culture medium (DMEM,
20% fetal calf serum, 1 × Glutamax, 1 × antibiotics and 50 μg/ml Endothelial Cell Growth Supplement (ECGS) (Sigma Chemical Co., USA)) and plated onto 0.1% gelatin coated dishes. Confluent
monolayers were incubated with 5 μg/ml DiI-Ac-LDL (Invitrogen) for 4 hours to mark endothelial cells, then scratched and monitored for cell migration in real time as described before8. Rate
of wound closure was calculated as the distance covered per min for around 200 cells at each wound margin. At least 30 cells per margin were selected randomly and monitored in real time
post-wounding for the tracking assay and analyzed for their directionality. CELL-MATRIX ADHESION ASSAY Control (NS) or _rudhira_ knockdown (KD) cells were plated at 2 × 104/well onto 96 well
plates that were pre-coated with collagen (20 μg/ml) or 0.1% gelatin and blocked with 0.5% BSA. After incubation of plated cells at 37 °C for the desired time, wells were washed in PBS,
fixed with 4% paraformaldehyde, stained with 5 mg/ml crystal violet, solubilised with 100 μl 2% SDS solution and absorbance measured at 550 nm. MATRIGEL INVASION ASSAY The assays and
quantitation were carried out as described before8. Briefly, control (NS) or _rudhira_ knockdown (KD) cells were serum-starved for 12 h and 20000 cells were plated onto the upper chamber of
transwell filter inserts with 8 μm pore size, 24-well format (Costar), pre-incubated with 0.03 mg/ml Matrigel for 4 h to initiate the assay. After the assay, the dye was extracted in
methanol and absorbance measured spectrophotometrically at 590 nm. SPHEROID SPROUTING ASSAY Endothelial spheroids were prepared as described26 with few modifications. 750 cells each of NS or
KD lines were taken for spheroid formation in a round-bottom non-adherent 96-well dish (Costar), in 1% CMC (carboxy methyl cellulose) in 10% FBS in DMEM. The spheroids formed were
transferred to collagen gels (Rat tail, Type I, Invitrogen) with a final concentration of 2.5 mg/ml. Gels were overlaid with 200 µl of 10% FBS in DMEM and the sprouting was monitored for 4–5
days. MICROARRAY SAMPLE PROCESSING AND DATA ANALYSIS E9.5 embryos of control and knockout littermates generated by crossing to _CMV-Cre_, were identified by embryonic tail genotyping.
Embryo and yolk sac were separated and total RNA extracted from each by TRIzol method. (For further details, see the supplementary Materials and methods.). All reported data are MIAME
compliant and raw data have been deposited in NCBI’s Gene Expression Omnibusand are accessible through GEO Series accession number
(http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE69204) a MIAME compliant database27. All other data generated or analyzed during this study are included in this published article (and
its Supplementary Information files). QUANTIFICATION AND STATISTICAL ANALYSES Statistical significance analyses were performed using One Way ANOVA in the Data Analysis package in Microsoft
Excel. p < 0.05 was considered significant. Statistical significance for frequency of embryos per genotype was calculated using chi-square test. REFERENCES * Huber, F., Boire, A., Lopez,
M. P. & Koenderink, G. H. Cytoskeletal crosstalk: when three different personalities team up. _Curr Opin Cell Biol_ 32, 39–47,
https://doi.org/10.1016/j.ceb.2014.10.005S0955-0674(14)00127-6 (2015). Article CAS PubMed Google Scholar * Chu, Y. W., Runyan, R. B., Oshima, R. G. & Hendrix, M. J. Expression of
complete keratin filaments in mouse L cells augments cell migration and invasion. _Proc Natl Acad Sci USA_ 90, 4261–4265 (1993). Article ADS CAS PubMed PubMed Central Google Scholar *
Helfand, B. T. _et al_. Vimentin organization modulates the formation of lamellipodia. _Mol Biol Cell_ 22, 1274–1289, https://doi.org/10.1091/mbc.E10-08-0699mbc.E10-08-0699 (2011). Article
CAS PubMed PubMed Central Google Scholar * Ivaska, J., Pallari, H. M., Nevo, J. & Eriksson, J. E. Novel functions of vimentin in cell adhesion, migration, and signaling. _Exp Cell
Res_ 313, 2050–2062, doi:S0014-4827(07)00145-010.1016/j.yexcr.2007.03.040 (2007). * Arden, S. D. _et al_. Molecular cloning of a pancreatic islet-specific glucose-6-phosphatase catalytic
subunit-related protein. _Diabetes_ 48, 531–542 (1999). Article CAS PubMed Google Scholar * Kim, G. W., Li, L., Gorbani, M., You, L. & Yang, X. J. Mice lacking alpha-tubulin
acetyltransferase 1 are viable but display alpha-tubulin acetylation deficiency and dentate gyrus distortion. _J Biol Chem_ 288, 20334–20350, doi:M113.46479210.1074/jbc.M113.464792 (2013). *
Yuba-Kubo, A., Kubo, A., Hata, M. & Tsukita, S. Gene knockout analysis of two gamma-tubulin isoforms in mice. _Dev Biol_ 282, 361–373,
doi:S0012-1606(05)00187-910.1016/j.ydbio.2005.03.031 (2005). * Jain, M., Bhat, G. P., Vijayraghavan, K. & Inamdar, M. S. Rudhira/BCAS3 is a cytoskeletal protein that controls Cdc42
activation and directional cell migration during angiogenesis. _Exp Cell Res_ 318, 753–767, doi:S0014-4827(12)00037-710.1016/j.yexcr.2012.01.016 (2012). * Siva, K., Venu, P., Mahadevan, A.,
Shankar, S. K. & Inamdar, M. S. Human BCAS3 expression in embryonic stem cells and vascular precursors suggests a role in human embryogenesis and tumor angiogenesis. _PLoS One_ 2, e1202,
https://doi.org/10.1371/journal.pone.0001202 (2007). Article ADS PubMed PubMed Central Google Scholar * Siva, K. & Inamdar, M. S. Rudhira is a cytoplasmic WD40 protein expressed in
mouse embryonic stem cells and during embryonic erythropoiesis. _Gene Expr Patterns_ 6, 225–234, S1567-133X(05)00069-410.1016/j.modgep.2005.06.002 (2006). * Nikpay, M. _et al_. A
comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. _Nat Genet_ 47, 1121–1130, https://doi.org/10.1038/ng.3396 (2015). Article CAS PubMed
PubMed Central Google Scholar * Copp, A. J. Death before birth: clues from gene knockouts and mutations. _Trends Genet_ 11, 87–93, doi:S0168-9525(00)89008-310.1016/S0168-9525(00)89008-3
(1995). * Rinkenberger, J. & Werb, Z. The labyrinthine placenta. _Nat Genet_ 25, 248–250, https://doi.org/10.1038/76985 (2000). Article CAS PubMed Google Scholar * Folkman, J.
Fundamental concepts of the angiogenic process. _Curr Mol Med_ 3, 643–651 (2003). Article CAS PubMed Google Scholar * Herbert, S. P. & Stainier, D. Y. Molecular control of
endothelial cell behaviour during blood vessel morphogenesis. _Nat Rev Mol Cell Biol_ 12, 551–564, https://doi.org/10.1038/nrm3176nrm3176 (2011). Article CAS PubMed PubMed Central Google
Scholar * Sun, X. & Feinberg, M. W. Regulation of endothelial cell metabolism: just go with the flow. _Arterioscler Thromb Vasc Biol_ 35, 13–15,
https://doi.org/10.1161/ATVBAHA.114.304869ATVBAHA.114.304869 (2015). * Bussolino, F., Mantovani, A. & Persico, G. Molecular mechanisms of blood vessel formation. _Trends Biochem Sci_ 22,
251–256, doi:S0968000497010748 (1997). * Morgan, J. T. _et al_. Nesprin-3 regulates endothelial cell morphology, perinuclear cytoskeletal architecture, and flow-induced polarization. _Mol
Biol Cell_ 22, 4324–4334, https://doi.org/10.1091/mbc.E11-04-0287mbc.E11-04-0287 (2011). Article CAS PubMed PubMed Central Google Scholar * Jin, Y. & Jakobsson, L. The dynamics of
developmental and tumor angiogenesis-a comparison. _Cancers (Basel)_ 4, 400–419, https://doi.org/10.3390/cancers4020400cancers4020400 (2012). Article Google Scholar * Wang, S. _et al_. The
endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. _Dev Cell_ 15, 261–271, doi:S1534-5807(08)00281-510.1016/j.devcel.2008.07.002 (2008). * Udan, R. S.,
Vadakkan, T. J. & Dickinson, M. E. Dynamic responses of endothelial cells to changes in blood flow during vascular remodeling of the mouse yolk sac. _Development_ 140, 4041–4050,
https://doi.org/10.1242/dev.096255dev.096255 (2013). Article CAS PubMed PubMed Central Google Scholar * Yagi, T. _et al_. A novel ES cell line, TT2, with high germline-differentiating
potency. _Anal Biochem_ 214, 70–76, S0003-2697(83)71458-210.1006/abio.1993.1458 (1993). * Schwenk, F., Baron, U. & Rajewsky, K. A cre-transgenic mouse strain for the ubiquitous deletion
of loxP-flanked gene segments including deletion in germ cells. _Nucleic Acids Res_ 23, 5080–5081, l50367 (1995). * Kisanuki, Y. Y. _et al_. Tie2-Cre transgenic mice: a new model for
endothelial cell-lineage analysis _in vivo_. _Dev Biol_ 230, 230–242, https://doi.org/10.1006/dbio.2000.0106S0012160600901064 (2001). Article CAS PubMed Google Scholar * Schlaeger, T.
M., Qin, Y., Fujiwara, Y., Magram, J. & Sato, T. N. Vascular endothelial cell lineage-specific promoter in transgenic mice. _Development_ 121, 1089–1098 (1995). CAS PubMed Google
Scholar * Korff, T. & Augustin, H. G. Integration of endothelial cells in multicellular spheroids prevents apoptosis and induces differentiation. _J Cell Biol_ 143, 1341–1352 (1998).
Article CAS PubMed PubMed Central Google Scholar * Edgar, R., Domrachev, M. & Lash, A. E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository.
_Nucleic Acids Res_ 30, 207–210 (2002). Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS We thank staff at the Jackson Laboratories, USA for inputs
on mouse breeding and maintenance; Aksah Sam for maintaining mouse stocks; Developmental Studies Hybridoma Bank, University of Iowa, USA for some antibodies; JNCASR Imaging facility, JNCASR
Animal Facility, NCBS Animal facility for access and Inamdar laboratory members for fruitful discussions. This work was funded by grants from the Department of Biotechnology, Government of
India (Sanction no. BT/PR11246/BRB/10/644/2008 dated 29.09.2009) the Wellcome Trust, UK (094879/B/10/Z) and intramural funds from Jawaharlal Nehru Centre for Advanced Scientific Research,
India. NCBS Animal facility is partially supported by DBT National Mouse Research Resource (NaMoR) grant (BT/PR5981/MED/31/181/2012;2013-2016). AUTHOR INFORMATION Author notes * Ronak Shetty
and Divyesh Joshi contributed equally to this work. AUTHORS AND AFFILIATIONS * Jawaharlal Nehru Centre for Advanced Scientific Research, Bangalore, India Ronak Shetty, Divyesh Joshi, Mamta
Jain, Jasper Chrysolite Paul, Ganesh Bhat, Poulomi Banerjee & Maneesha S. Inamdar * RIKEN Center for Life Science Technologies, Kobe, Japan Takaya Abe & Hiroshi Kiyonari * Bionivid,
Kasturi Nagar, Bangalore, India Madavan Vasudevan * National Centre for Biological Sciences, Bangalore, India K. VijayRaghavan * Institute for Stem Cell biology and Regenerative Medicine
(inStem), Bangalore, India Maneesha S. Inamdar Authors * Ronak Shetty View author publications You can also search for this author inPubMed Google Scholar * Divyesh Joshi View author
publications You can also search for this author inPubMed Google Scholar * Mamta Jain View author publications You can also search for this author inPubMed Google Scholar * Madavan Vasudevan
View author publications You can also search for this author inPubMed Google Scholar * Jasper Chrysolite Paul View author publications You can also search for this author inPubMed Google
Scholar * Ganesh Bhat View author publications You can also search for this author inPubMed Google Scholar * Poulomi Banerjee View author publications You can also search for this author
inPubMed Google Scholar * Takaya Abe View author publications You can also search for this author inPubMed Google Scholar * Hiroshi Kiyonari View author publications You can also search for
this author inPubMed Google Scholar * K. VijayRaghavan View author publications You can also search for this author inPubMed Google Scholar * Maneesha S. Inamdar View author publications You
can also search for this author inPubMed Google Scholar CONTRIBUTIONS M.S.I. conceived of the project and directed the work. M.S.I., R.S., D.J., J.C.P., M.J., G.B., P.B., T.A. and H.K.
designed and performed experiments. R.S., M.V., M.S.I., D.J. analyzed transcriptome data. M.S.I., R.S., D.J., M.V., K.V.R. wrote the manuscript. CORRESPONDING AUTHOR Correspondence to
Maneesha S. Inamdar. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER'S NOTE: Springer Nature remains neutral with
regard to jurisdictional claims in published maps and institutional affiliations. ELECTRONIC SUPPLEMENTARY MATERIAL SUPPLEMENTARY INFORMATION 41598_2018_24014_MOESM2_ESM.XLS S2
41598_2018_24014_MOESM3_ESM.XLS S3 41598_2018_24014_MOESM4_ESM.DOC S4 41598_2018_24014_MOESM5_ESM.XLSX S5 41598_2018_24014_MOESM6_ESM.XLS S6 RIGHTS AND PERMISSIONS OPEN ACCESS This article
is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give
appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in
this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative
Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a
copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Shetty, R., Joshi, D., Jain, M. _et al._ Rudhira/BCAS3
is essential for mouse development and cardiovascular patterning. _Sci Rep_ 8, 5632 (2018). https://doi.org/10.1038/s41598-018-24014-w Download citation * Received: 14 November 2017 *
Accepted: 22 March 2018 * Published: 04 April 2018 * DOI: https://doi.org/10.1038/s41598-018-24014-w SHARE THIS ARTICLE Anyone you share the following link with will be able to read this
content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative