Insights into the mechanism of the pik3ca e545k activating mutation using md simulations
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Phosphoinositide 3-kinase alpha (PI3Kα) is involved in fundamental cellular processes including cell proliferation and differentiation and is frequently mutated in human
malignancies. One of the most common mutations is E545K, which results in an amino acid substitution of opposite charge. It has been recently proposed that in this oncogenic charge-reversal
mutation, the interactions between the protein catalytic and regulatory subunits are abrogated, resulting in loss of regulation and constitutive PI3Kα activity, which can lead to
oncogenesis. To assess the mechanism of the PI3Kα E545K activating mutation, extensive Molecular Dynamics simulations were performed to examine conformational changes differing between the
wild type (WT) and mutant proteins as they occur in microsecond simulations. In the E545K mutant PI3Kα, we observe a spontaneous detachment of the nSH2 PI3Kα domain (regulatory subunit,
p85α) from the helical domain (catalytic subunit, p110α) causing significant loss of communication between the regulatory and catalytic subunits. We examine the allosteric network of the two
proteins and show that a cluster of residues around the mutation is important for delivering communication signals between the catalytic and regulatory subunits. Our results demonstrate the
dynamical and structural effects induced by the p110α E545K mutation in atomic level detail and indicate a possible mechanism for the loss of regulation that E545K confers on PI3Kα. SIMILAR
CONTENT BEING VIEWED BY OTHERS STRUCTURAL BASIS OF PHOSPHATIDYLINOSITOL 3-KINASE C2Α FUNCTION Article Open access 07 March 2022 MICROSECOND-TIMESCALE MD SIMULATION OF EGFR MINOR MUTATION
PREDICTS THE STRUCTURAL FLEXIBILITY OF EGFR KINASE CORE THAT REFLECTS EGFR INHIBITOR SENSITIVITY Article Open access 16 April 2021 THE STRUCTURAL BASIS OF PTEN REGULATION BY MULTI-SITE
PHOSPHORYLATION Article 08 October 2021 INTRODUCTION Phosphoinositide 3-kinase alpha (PI3Kα) is a lipid kinase that plays a crucial role in cell processes involving growth and survival1.
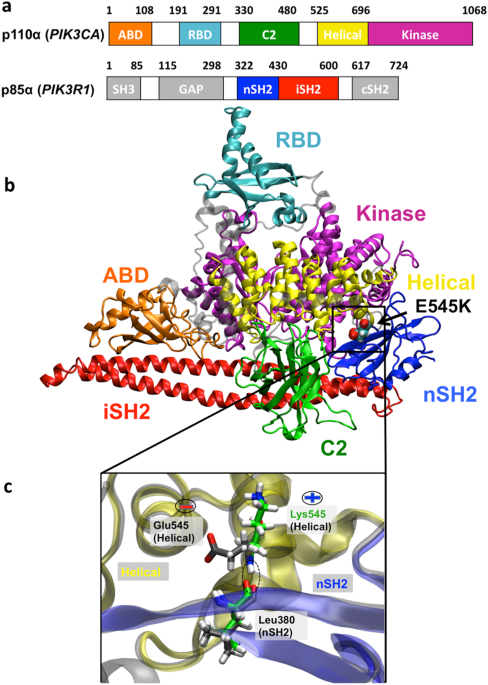
PI3Kα is comprised of a catalytic subunit, p110α, encoded by the _PIK3CA_ gene, and a regulatory subunit, p85α, encoded by the _PIK3R1_ gene (Fig. 1). Somatic missense mutations of p110α
have been identified as the most common mutations associated with a variety of human cancer types2,3,4,5,6,7,8. The two most common cancer-linked mutations are H1047R and E545K, located in
the kinase and helical domains of p110α, respectively6,9. These hotspot mutations transform cells _in vitro_10 and enhance tumorigenicity in transgenic mouse models11. Currently, several
PI3Kα inhibitors are under development and in clinical trials12. Alone or in combination with other drugs, PI3Kα inhibitors prevent tumor growth particularly in estrogen receptor alpha
(ERα)-positive breast cancers, but depending on the type of the PI3Kα mutation different therapeutic regimes must be followed13. Structural and functional studies of the PI3Kα wild type (WT)
and hotspot mutants are of paramount importance for understanding the effect of oncogenic mutations on the constitutive lipid kinase activity of _PIK3CA_ (p110α subunit) and consequently
for the discovery of mutant-specific inhibitors. The crystal structure elucidation of PI3Kα WT14,15,16,17 and H1047R mutant18 has enabled the mechanistic understanding of PI3Kα
overactivation by the H1047R mutant. By using a combination of Molecular Dynamics (MD) simulations and experiments, it was shown that the H1047R mutation, which is located in the kinase
domain of PI3Kα, abolishes the auto-inhibitory role of the C-terminal tail of the protein, enhances protein-membrane binding, and alters the conformation of H917, a critical residue for ATP
hydrolysis19,20. Although a crystal structure bearing the PI3Kα E545K mutation, which is located in the helical domain of PI3Kα, is not yet available, several studies suggest that this
mutation leads to elevated kinase activity by an entirely different mechanism than H1047R. Moreover, it has been shown in biochemical experiments that when both mutations are introduced they
act independently and synergistically, indicating that the molecular mechanisms for the mutation-induced gain of function in helical and kinase domain mutations are complementary and
reciprocal21. Helical domain mutants (such as E545K) lead to activation by abolishing the inhibitory contact of p85α that regulates PI3Kα activation (such mutants are p85α independent),
however, they still need to interact with RAS to reach the membrane. On the other hand, while kinase domain mutants (such as H1047R) are active in RAS absence, they require the interaction
with the regulatory subunit p85α21. Early studies have indicated that E545K acts by altering the p110α-p85α interface22,23. In a low activity state, the nSH2 and iSH2 domains of the
regulatory subunit p85α stabilize p110α and inhibit its enzymatic activity. In one of these inhibitory contacts, the nSH2 domain of p85α interacts strongly with the helical domain of p110α
and stabilizes the inactive state of PI3Kα. This contact is physiologically released by phosphotyrosine (pY)-containing peptides, which bind at the nSH2-helical interface and activate the
enzyme (pY-activated state of the WT)24,25. The E545K charge reversal mutation in the helical domain of p110α is located exactly at the position where pY-peptides bind nSH2. Later studies
also suggest that the E545K mutant increases lipid kinase activity by disrupting the nSH2-helical interface, mimicking the pY-activated state of the protein21,26,27,28. This mechanism has
been further supported by biochemical studies investigating the kinetics of PI3Kα activation for the WT and oncogenic mutant (E545K, H1047R) proteins27,29. Both mutants show a 2-fold
increase in lipid kinase activity compared to the activated WT29. However, contrary to H1047R, the E545K mutant cannot be further activated in the presence of growth factor stimulation
(pY-peptides), suggesting that this mutant activates p110α through the same mechanism as the binding of pY-peptides27. Nevertheless, so far, there is no crystal structure of the mutant E545K
available to provide structural evidence for this mechanism. A recent hydrogen/deuterium exchange mass spectrometry (HDX-MS) study gave the first insight into the structural mechanism of
activation of PI3Kα caused by the E545K mutation30. Burke _et al_. showed that the E545K helical mutation enhances three out of the four distinct events that occur during the physiological
transition of the WT PI3Kα from an inactive to an active state. These three events require breaking of the nSH2-helical interface, disruption of the iSH2-C2 interface, and movement of the
ABD domain relative to the rest of the catalytic subunit30. Although the experiments capture the dynamic nature of the enzyme activation, the order with which these events are taking place
remains unclear. MD simulations have been successfully used for the identification of intermediate structures during the activation pathway of various kinases19,31. Conventional MD32,33,34
or biased, enhanced sampling methodologies35,36,37 have been extensively used to study the mechanism of constitutive activation of various protein kinases as well as the mechanism of action
of anti-cancer drugs35,37,38. A recent MD study of oncogenic mutations of PI3Kα suggests that all tumor-associated mutants weaken p110α-p85α interactions by increasing positional
fluctuations of nSH2 (p85α) domain39. However, the limited sampling of these simulations could not allow the observation of significant conformational changes. Another recent study of E545K
and E542K mutants of PI3Kα, involving docking of catalytic (p110α) and regulatory (p85α) subunits and subsequent MD simulations, indicates fewer interactions of p110α-p85α in the mutants
compared to the native structure40. In this study, a connection between nSH2 (p85α) flexibility and helical mutations (E545K, E542K) was observed, however, it remains unclear how this effect
could eventually lead to nSH2 detachment and allosteric regulation of the enzyme activity, perhaps due to the approximation of docking the two subunits and not using the crystal structure
in the simulations. In the present study, we performed microsecond-long simulations of the full-length WT and E545K p110α/niSH2 heterodimer to investigate the structural and dynamical
changes induced by the mutation through unbiased MD simulations. For the first time, we have been able to follow in atomic-level detail the unforced detachment of nSH2 (p85α) from the
helical (p110α) domain in the PI3Kα mutant form, monitoring the series of events leading to this disruption. The molecular mechanism that emerges from our study could explain how the nSH2
domain detaches gradually from the helical one, resulting in loss of this inhibitory contact that can eventually lead to overactivation of the enzyme. We also investigated the allosteric
network connecting key residues in the WT and mutant proteins and show that a cluster of residues around 545 is critical for delivering communication signals between the catalytic and
regulatory subunits. Our simulations are in agreement with HDX experimental data and allow us to provide atomic-detail insights into the mechanism of the _PIK3CA_ E545K activating mutation
by monitoring structural and dynamical elements of the WT and mutant proteins. RESULTS SPONTANEOUS DETACHMENT OF THE NSH2 (P85Α) DOMAIN FROM THE P110Α SUBUNIT DISRUPTION OF NSH2-HELICAL
INTERFACE IN THE E545K MUTANT Calculation of the Root Mean Square Deviation (RMSD) with respect to the initial structure shows that a significant conformational change takes place in one of
the PI3Kα E545K mutant simulations (Mutant run 1, Supplementary Fig. S1). Also, as seen in Supplementary Fig. S2, the Root Mean Square Fluctuation (RMSF) of the nSH2 domain is significantly
increased in the mutant compared to the WT. A detailed analysis of the conformational change shows that it is attributed mostly to the movement of the nSH2 domain (p85α) relatively to the
helical one (p110α) (Fig. 2). Initially, the nSH2-helical domains are in close contact forming several hydrogen bonds (closed-state) as in the WT crystal structure (a detailed description of
the polar network interactions is given at the end of this section). Destabilization of this interaction network at the nSH2-helical interface in the mutant protein, triggers a significant
conformational change. nSH2 gradually moves away and detaches from the helical domain in accordance with previous HDX experiments30 and structural studies18. The movement of nSH2 relative to
the helical domain is clearly seen in Fig. 2a, which shows the distance between the center of mass of the two domains increasing upon the detachment. Notice, that after the detachment (t =
500 ns, dotted lines in Fig. 2) nSH2 approaches the helical domain for a short period (transient attachment, t = 670 ns), but then again moves away (t = 800–900 ns, Fig. 2a). A step-wise
character of the disruption is revealed by examining the time series of the total number of hydrogen bonds formed between nSH2-helical (Fig. 2d). At t = 200 ns, the total number of hydrogen
bonds between nSH2-helical decreases from 4 to 2 (Step 1). The decrease in the total number of hydrogen bonds results in increasing fluctuations of the distance between the centers of mass
of nSH2-helical domains (Fig. 2a). At t = 500 ns (Step 2) an almost complete nSH2-helical hydrogen bond loss takes place as can be seen in Fig. 2d. The nSH2-helical hydrogen bond network
disruption leads to the actual detachment, where a 5 Å increase in the nSH2-helical center-of-mass distance is observed (open-state). A transient re-attachment of nSH2 occurs at around 670
ns due to the formation of a hydrogen bond between the nSH2-helical domains (Step 3), which, however, is lost at around 800 ns and the nSH2 detaches from the helical domain at the end of the
simulation run (Step 4). The total number of hydrogen bonds between nSH2-helical domains is consistently lower in all mutant simulations with respect to the WT (Supplementary Figure S3). On
the contrary, the nSH2-helical distance and the total number of hydrogen bonds between the two domains (Fig. 2a, d and Supplementary Figure S3) in the WT protein remain constant indicating
a stable nSH2-helical contact. For a detailed explanation of the sequence of events observed upon detachment and the role of the charge reversal, see the Supplementary Information. A video
with the trajectory of the detaching nSH2 domain from the helical domain can be seen in Supplementary Video S1. To better understand the hydrogen bond disruption during the nSH2-helical
detachment, we have calculated the time series of nSH2-helical hydrogen bonds, which are discussed below. The percentage of occupancy of inter-domain hydrogen bonds in the vicinity of the
E545K mutation for all simulated systems can be found in Supplementary Tables S1-S3. Here, we do not focus on the total number of hydrogen bonds (that could involve the formation of
transient hydrogen bonds between residues at the nSH2-helical interface), but rather on the formation and disruption of persistent hydrogen bonds between specific interfacial residues.
Breaking of these hydrogen bonds could possibly trigger the observed conformational change. As seen in Fig. 3a and Supplementary Table S1, the L380(nSH2)-K545(helical) and
K382(nSH2)-Q546(helical) hydrogen bonds in the Mutant run 1 simulation, are the first to be disrupted as a result of the weakening of the bond K379-545K, which ensues after the
charge-reversal mutation (Supplementary Table S3) (Step 1). The only remaining hydrogen bond in the vicinity of K545 is R340(nSH2)-E542(helical), which keeps the two domains in close
contact. The disruption of this last strong inter-domain hydrogen bond interaction (Step 2) initiates the movement of nSH2 away from the helical domain. The two domains associate transiently
again due to the formation of the critical hydrogen bond R340(nSH2)-E542(helical) (Fig. 3a) (Step 3), but then the loss of this hydrogen bond leads again to the dissociation of the two
domains (Step 4). In a previous experimental study, it was shown that the p85ni-R340E mutant did not inhibit the WT p110α, and it was suggested that R340 is involved in an inhibitory contact
with the catalytic subunit25. Apparently, E542 acts as the last residue to preserve the nSH2-helical inhibitory contact. It should be noted that a persistent interaction of E542
(R358(nSH2)-E542(helical)) in the WT, is almost completely absent in the mutant E545K simulation (Fig. 3a,d and Supplementary Tables S2 and S3). In fact, we show in our allosteric analysis
(see the “Dynamical Network Analysis” Section) that the communication between R358(nSH2)-E542(helical) is weakened as an effect of the mutation E545K. The importance of the E542(helical)
residue in preserving the inhibitory role of nSH2 is well known. Biochemical analysis of a double E545K-E542K mutant has revealed a simple additive behaviour in the enzyme overactivation,
suggesting that the two helical mutations act through the same mechanism, which involves the release of the nSH2-helical inhibitory contact21. EFFECT OF E545K ON THE NSH2-C2 AND NSH2-KINASE
INTERFACES The detachment of nSH2 from the helical domain in the mutant protein also alters the interactions of nSH2 with the neighboring C2 and kinase domains of the catalytic p110α
subunit. As seen in Fig. 2b, the center of mass distance between nSH2 and C2 is initially 3.3 nm, but after the detachment it oscillates between 3.3 ± 0.1 and 3.8 ± 0.1 nm. In contrast, in
the case of the WT run 1 simulation, the center of mass distance between nSH2 and C2 remains stable at 3.2 ± 0.1 nm. In all mutant simulations (Table 1 and Supplementary Fig. S3), the nSH2
domain approaches the kinase domain starting from 4.8 ± 0.1 nm and reaching to a mean value of 4.4 ± 0.1 nm. In the WT simulations, the nSH2 – kinase center of mass distance remains stable
at 4.3 ± 0.1 nm (Fig. 2). Despite the fact that the relative positioning of nSH2 changes, the total number of hydrogen bonds between nSH2-C2 and nSH2-kinase domains is constant throughout
the simulation (Fig. 2e, f) and is not affected by the detachment. Overall, the total number of hydrogen bonds between nSH2-C2 and nSH2-kinase domains ranges between 2–4 and 4–6,
respectively, for all simulated systems (Fig. 2, Supplementary Fig. S3 and Supplementary Table S3). Therefore, even though nSH2 has lost contact with the helical domain of the p110α
catalytic subunit during detachment, it maintains essential polar contacts with the other two neighboring domains (C2, kinase) that presumably keep it from moving far away from the complex.
nSH2 contacts the kinase domain with its 339–347 helix, which interacts with helix α10 of the C-lobe of the kinase domain. We observe that during the detachment, the 339–347 helix serves as
a hinge around which nSH2 rotates and detaches from the helical domain (Fig. 4). As seen in Fig. 4e, the angle φ (shown in Fig. 4d) that is formed between the center of mass of residue
E545K, the center of mass of helix 339–347, and that of helix 400–410 (which is located in the solvent exposed part of the nSH2 domain), ranges from 60°, at the beginning of the simulation
(Fig. 4a, c), to 90° upon the detachment (Fig. 4b, d). According to HDX-MS data, “the one area in the nSH2 that shows decreased HDX in p110α compared to p110β and p110δ is the helix A
(residues 339–347). This may explain why the nSH2 more strongly inhibits the p110α subunit compared to p110β, and p110δ. Although there is no crystal structure of the p110β or p110δ
interacting with the nSH2, the HDX-MS suggests that there are conformational differences with respect to p110α that result in more protection of the nSH2, likely due to it making more
extensive contacts in the p110α/p85 complex”41. Indeed, our simulations show that this interaction persists until the end of our simulation, and we may hypothesize that the detachment
described in the current work constitutes the first step for the complete dissociation of nSH2 from its neighboring counterparts, which will lead to constitutive relief of inhibition. LOSS
OF COMMUNICATION BETWEEN FUNCTIONAL DOMAINS It is well known that kinases have a dynamic infrastructure that can be disrupted by single point mutations, resulting in allosteric regulation of
enzyme activity42,43,44,45 even in the absence of large conformational changes. In our case, where a significant conformational change takes place, we expect to see differences in the
communication between domains of PI3Kα in the WT and E545K mutant proteins, indicating a possible mechanism for allosteric regulation of the enzyme activity controlled by E545K. Comparison
of the RMSF of the E545K mutant simulation, where nSH2 detaches (open-state), with the WT (closed-state) shows that there are specific regions in functional domains that are more flexible in
the mutant open-state. As seen in Supplementary Fig. S4, residues in certain regions of nSH2, helical, and iSH2 domains that are in proximity to the disrupted, open-state nSH2-helical
interface show enhanced flexibility that is increased in the order of RMSF = 0.1–0.2 nm compared to the closed-state (Supplementary Fig. S5 and Table S4). The locations of the increased
flexibility regions of the open-state protein are in agreement with HDX experiments30 (Supplementary Fig. S4). Specifically, increased fluctuations are observed for residues 374–424 that
constitute a β-sheet and an α-helical part of nSH2 that is in close contact with the E545K mutation site and for residues 541–553 that include the mutation site on the helical domain
(Supplementary Fig. S4). Furthermore, residues 442–452 (iSH2) of the regulatory subunit are also more flexible in the mutant open state compared to the rest of the simulation runs that are
sampling the closed state (Supplementary Fig. S5 and Table S4). This flexibility could be expected as residues 442–452 of iSH2 are connected by chain continuity with the detached nSH2
domain. There are two additional regions, residues 374–380 of the C2 domain and residues 784–794 of the kinase domain that show increased flexibility in the mutant open-state. However, these
specific regions are implicated neither in inter-domain nor in functionally important interactions. MAPS OF DISTANCE FLUCTUATIONS In order to assess the effect of the E545K mutation on the
plasticity and coordination between domains, we have calculated the distance fluctuation (DF) maps for the Cα atoms of the nSH2-helical, nSH2-C2 and nSH2-kinase domains46 (see Methods). The
results are presented in Fig. 5 for the WT (closed-state) and the mutant (open-state) proteins. Values of distance fluctuations that are lower than 0.3 Å2 and correspond to pairs of atoms of
nSH2-helical, nSH2-C2, and nSH2-kinase domains, indicate rigid body-like motions that could propagate strong allosteric communication between these domains. Therefore, as seen in the Mutant
run 1 simulation (open-state, Fig. 5), the detachment of nSH2 results in significant distance fluctuations, weakening the communication between nSH2-helical and nSH2-C2 domains.
Interestingly, although nSH2 seems to physically approach the kinase domain (Fig. 2c) the communication between the two is also distorted with respect to the WT (Fig. 5). The conformational
plasticity has been also calculated for mutant replica runs (Table 1), however, it does not show a significant change compared to the WT protein (Supplementary Fig. S6). DYNAMICAL NETWORK
ANALYSIS In allosteric communication, a signal is propagated over a long distance through the protein structure from one site to another. It has been argued that allosteric communication in
proteins relies upon networks of collective, rigid body motions and residue-residue contact motions47,48,49. To understand the role of residue E545K in allosteric regulation within PI3Kα, we
have constructed network models obtained from our MD simulations. For our analyses, we used the Dynamical Network Analysis method as implemented in the program VMD50,51 (see Methods section
and the SI for a detailed explanation). Based on this model, allosteric signals are dependent on positional correlations of protein residues, and correlated motion is used to generate a
weight of the signal transmitted through two residues. Apart from identifying allosteric pathways, the method also applies hierarchical clustering using the Girvan-Newman algorithm to
cluster residues whose motion is highly correlated in so-called “communities” that are highly intra-connected but loosely inter-connected (see SI for more information)52. The communication
between different communities passes through specific residue interactions that form critical edges. A detailed analysis of the inter-domain communication network has been performed for the
WT (closed-state) and mutant (open-state) using the dynamical network analysis method (see Methods section). As can be seen in Fig. 6a, the clustering of residues in “communities” largely
reflects the organization of the functional domains that has been proposed for PI3Kα, which is also schematically depicted in Fig. 1. While the PI3Kα domains are very well characterized by
the communities calculated from the network analysis, some residues act at the interface between domains and propagate their motion within the allosteric network of the protein, and thus,
depending on their positional correlation they may be categorized in an adjacent PI3Kα domain (Fig. 6b, c). For the WT protein, the majority of the paths connecting residues between the nSH2
and helical domains pass through the edge defined by T544(helical community) and E547(nSH2 community) residues (Fig. 6b). Moreover, the nSH2 domain is additionally connected to the helical
domain, through residues L540, P539, which belong to the helical community, and E542, R537, which belong to the nSH2 community, according to the network analysis. Clearly, the residues
around the hotspot mutations E542/E545 play a critical role in the communication of the helical domain with the nSH2 domain (for more details about the edges between nSH2 and helical
domains, the reader is referred to Supplementary Table S5). However, this communication is completely lost upon the detachment of nSH2 from the helical (Fig. 6c). One of the key interactions
between nSH2 and helical domains that is almost immediately lost in the majority of the E545K mutant simulations, is the R358-E542 salt bridge (Fig. 3a and Supplementary Table S3). Our
network model indicates that the communication between 545K and E542 is preserved, while the communication between 545K and R358 is significantly reduced with respect to the WT (Fig. 7). The
optimal path of communication between 545K and R358 is substantially longer compared to E545 – R358 (Fig. 7b-d). For the R358 – E542 salt bridge, the position of R358 is maintained close to
E542 through a motional correlation with E545 (Fig. 7b), but when E545 is mutated to 545K, the weakening of the communication between E545 and R358 (Fig. 7c, d) leads to the breaking of the
R358-E542 salt bridge. The communication between nSH2-C2 domains is also significantly distorted in the mutant open-state. Although upon the nSH2-helical detachment the nSH2-C2 interface is
largely affected, based on the maps of distance fluctuations (Fig. 5), one critical edge involving residue D349 nSH2 domain remains (Fig. 6), which may preserve the communication between
the two domains. Another domain of the regulatory subunit, iSH2, directly interacts and possibly controls the kinase, C2, and ABD domains of the catalytic subunit. However, analysis of the
communication network between these domains has shown that they are not significantly affected by the conformational change taking place at the helical-nSH2 interface. Interestingly,
iSH2-kinase communication takes place through the activation loop of kinase and is highly conserved even after nSH2 detachment (Fig. 6). PROPOSED MECHANISM FOR P85Α INHIBITION
NSH2-ACTIVATION LOOP INTERACTIONS Although the kinase domain of the catalytic region of PI3Ks is highly conserved, members of Class I PI3K exhibit high lipid substrate specificity,
determined by residues on the activation loop (res. 933–958) (see Fig. 8a). Amino acid substitutions and structural modelling have shown that the positively charged amino acids of the
activation loop are crucial for the selective binding and phosphorylation of phosphatidylinositol (PI) and its phosphorylated derivative phosphatidylinositol 4,5-biphosphate (PIP2)53.
Indeed, a recent crystallographic structure (PDB ID 4OVV) of PI3Kα in complex with a PIP2 mimetic, reveals that PIP2 binds very close to the activation loop and forms a salt-bridge or
water-mediated hydrogen bond with K941 of the activation loop (Fig. 8b)15. Moreover, the activation loop forms a salt-bridge between K948 (kinase) and E342 (nSH2) (Supplementary Table S3).
The position of PIP2 reveals that the previously suggested base for ATP hydrolysis, H917, is too far away from the lipid substrate. Therefore, another base (possibly H936), which is closer
to the lipid substrate, should be able to deprotonate the 3′-hydroxyl of PIP2. A schematic representation of the area around the activation loop and the lipid substrate binding pocket is
given in Fig. 8b-d. Based on this crystallographic structure (PDB ID 4OVV), Miller and co-workers have proposed a possible mechanism by which nSH2 inhibits the catalytic activity15. They
suggest that the interactions that nSH2 forms with the activation loop regulate its position and keep the latter in its inactive conformation, away from the lipid substrate binding site. The
detachment of nSH2 (p85α) from the helical (p110α) domain should disrupt the interactions between nSH2 and the activation loop, and allow the latter to adopt an active-like conformation, in
which it will be positioned in the vicinity of the lipid substrate binding site. We find in our simulations that nSH2 forms persistent hydrogen bonds with the activation loop in the WT
PI3Kα protein (Fig. 3). More specifically, the E342(nSH2)-K948(activation loop) hydrogen bond is formed both in WT and mutant proteins, while in the WT protein an additional
E341(nSH2)-Y947(activation loop) hydrogen bond is formed. We observe a constant and stable interaction of nSH2 with the activation loop in all MD runs of the WT protein, where nSH2 is in
contact with the helical domain (Fig. 3f and Supplementary Table S3). In accordance with theoretical predictions based on structural studies15,18, the E342(nSH2)-K948(activation loop) bond
in the mutant breaks during nSH2 detachment from the helical domain resulting in a total loss of contact and communication between nSH2 and activation loop. Although the nSH2-activation loop
interaction is diminished in the mutant open-state (Mutant run 1), we do not observe any significant conformational change in the activation loop that could be attributed to the detachment
of nSH2 (Fig. 8c, d). This is also confirmed by the time series of distances between Cα atoms of key residues of the two domains for all simulated systems (Supplementary Fig. S7). Therefore,
the positioning of essential residues for binding and deprotonation of the lipid substrate (PIP2) remains unchanged even in the absence of nSH2-activation loop interactions. We offer below
an explanation why this may be the case. ISH2-ACTIVATION LOOP INTERACTIONS The dynamics of the activation loop is not significantly altered after nSH2 detachment (Supplementary Fig. S5,
activation loop). The average value of RMSF of the activation loop in the WT (simulations: WT run 1, replica 1) ranges between 0.118 ± 0.039 nm − 0.124 ± 0.031 nm (Supplementary Table S4).
The same value becomes 0.107 ± 0.027 nm in the mutant open-state simulation (Supplementary Table S4). The low RMSF value of the mutant open state indicates that the activation loop does not
experience a conformational change. We observe that while the interaction with the nSH2 domain is lost (Supplementary Fig. S8a, b), the activation loop enhances its interactions with the
third helix of the iSH2 domain (p85α residues 587–598), iα3, especially after the detachment of the nSH2 domain (after 500 ns, Supplementary Fig. S8c, d). In the WT crystal structure15, this
interface is mediated by hydrophobic contacts between L598 (iSH2, p85α) and F945 (activation loop, p110α) and a hydrogen bond between Q591 (iSH2, p85α) and K948 (activation loop, p110α). Ιn
the mutant E545K these interactions are enhanced. iSH2 attracts the activation loop through mainly hydrophobic interactions (iSH2 N595 interacts with activation loop Y947, iSH2 K592 with
activation loop Y947, iSH2 W597 with activation loop K944, iSH2 L598 with activation loop K941, iSH2 L594 with activation loop F945, iSH2 Q591 with activation loop F945). One hydrogen bond
is formed between iSH2 W597 and the activation loop K944, and one salt bridge is formed between the C-terminal carboxylic acid of iSH2 L598 and the side chain of the activation loop K944. We
should note finally, that the activation loop does not become more solvent exposed after nSH2 detachment (Supplementary Fig. S8e, f), in accordance with HDX-MS data, in which the deuterium
exchange of the region corresponding to the activation loop remained unaffected upon the E545K mutation compared to the WT41. FAST KINETICS AND INTRINSIC FLEXIBILITY OF NSH2 The detachment
of the nSH2 from the helical domain is observed only in one of the eight mutant simulations, which restricts the statistics of the specific event. However, the total number of nSH2-helical
hydrogen bonds in all mutant simulations is lower than in the WT runs (Fig. 2 and Supplementary Fig. S3). In the WT simulations, the total number of nSH2-helical hydrogen bonds is ~6, while
in the mutant runs it fluctuates between 2 and 4 hydrogen bonds. Interaction energies between nSH2 and helical domains are plotted in Supplementary Fig. S9. It is clearly seen that the
nSH2-helical polar interactions are less favorable in the mutant simulations. Nevertheless, the distance fluctuations maps (Supplementary Fig. S6) do not show any significant change in the
communication network compared to the WT runs. We could argue that the less favorable nSH2-helical interactions in the E545K mutant replicate simulations could eventually lead to the
disruption of the interface resulting in the detachment of nSH2 from the helical. Large conformational changes in protein kinases take place over a timescale of microseconds to
milliseconds54. However, we have been able to observe the rare event of the spontaneous detachment of nSH2 from the helical domain in the E545K mutant, on a much shorter timescale of ~500
ns. The relatively fast kinetics of this conformational change could be associated with the intrinsic disorder of the nSH2 domain. It is not a coincidence that most of the crystal structures
of WT PI3Kα lack diffraction data of the nSH2 domain. The first data indicating the structure and positioning of nSH2 in the p110-niSH2 complex were given by the H1047R mutant crystal
structure18. Also, very recently a specific p110α-p85α niSH2 fusion construct (PDB IDs 4L1B, 4L2Y)16 was employed in order to capture the nSH2 domain. The free p110α-p85α niSH2 complex that
we are using in this study (PDB ID 4OVU)15 has shown a considerable degree of ordering in the nSH2 domain. However, even in this structure, the intrinsic disorder of nSH2 is still evident in
the relatively high B factors15 of nSH2 and the region of iSH2 in the proximity of nSH2. Expensive Metadynamics calculations are required to increase the sampling and provide valuable
information about intermediate structures and energetic barriers. However, these calculations are still beyond the current accessible computational time for such large systems (500 K atoms)
to reach convergence and provide accurate insights. DISCUSSION Biochemical and structural studies suggest that the E545K oncogenic mutant mimics and enhances the physiological activation of
the PI3Kα kinase by releasing nSH2 (p85)-helical(p110) inhibitory contacts21,27,30. Indeed, our simulations indicate a significant reduction in polar interactions at the nSH2-helical
interface in the case of the mutant protein. Interestingly, a complete nSH2 detachment from the helical domain is observed in one out of the eight simulation runs of the mutant. The
alteration of the polar contact network around the mutant residue 545K and the breaking of crucial hydrogen bonds between nSH2 and helical domains, lead to the detachment of the two domains
that can be observed through a stepwise mechanism (Fig. 9). A critical step towards this significant conformational change is breaking of two specific inter-domain hydrogen bonds
(L380(nSH2)-E545(helical) and K382(nSH2)-Q546(helical)) that are present in all PI3Kα complexes (Fig. 2) (Step 1). Subsequently, an interplay between a partially and fully detached
nSH2-helical complex is observed where the role of residue E542 (helical) seems to be important (Step 2). The two domains associate transiently again as seen in Fig. 9 (Step 3), but then
loss of the R340(nSH2)-E542(helical) critical salt bridge leads to the complete dissociation of the two domains (Fig. 2) (Step 4). Although the total polar contacts of the nSH2-C2 and
nSH2-kinase interfaces remain unaffected, monitoring the distance fluctuations between nSH2 and the helical, C2, and kinase domains revealed that the communication between them is weakened.
Moreover, the detachment of the helical-nSH2 domains leads to enhanced flexibility of certain areas of the nSH2, helical and iSH2 domains, in agreement with HDX experiments30 (Supplementary
Fig. S4). To further assess the allosteric network of the two proteins, we used the dynamical network analysis method, which indicates that opening of the nSH2-helical interface results in a
significant loss of communication between the regulatory and catalytic subunits of the kinase. Moreover, we found that communication signals are transported from the helical to the nSH2
domain through a cluster of helical domain residues R537, P539-E542, T544, and E547. In the WT protein, the nSH2 domain locks the activation loop in an inactive conformation via a
salt-bridge between K948 (p110α) and E342 (p85α). At the same time, K948 is also locked by an interaction with Q591 of the iSH2 domain. The mutation E545K at the helical-nSH2 interface
causes a conformational change that breaks the K948 (p110α) – E342 (p85α) interaction with the activation loop, resulting in loss of communication between nSH2 and the activation loop of the
catalytic subunit. The loss of contact between the activation loop and nSH2 is accompanied by an enhanced interaction between the activation loop and the iSH2 domain, which maintains an
inhibitory role for the activation loop. Loss of contact with iSH2 would also be required for activation; this may be induced by the approach of the high negative charge of PIP2 in the
proximity of the positive activation loop (Fig. 8), which is lysine and arginine-rich. We postulate that the release of this inhibitory contact between p85α and the activation loop would
allow the activation loop to adopt an active conformation. We have observed here for the first time in atomic-level detail the spontaneous detachment of the nSH2-helical domains in the E545K
mutant and provide a possible mechanism for the loss of regulation that E545K confers based on our simulation results. Insights gained from this study could assist the design of
mutant-specific PI3Kα inhibitors that exploit the altered conformation of the mutant with respect to the WT protein. METHODS The entire crystal structure of the human WT PI3Kα (PDB ID:
4OVU)15 was used for the simulations. A schematic representation of PI3Kα heterodimer with the p110α catalytic and p85α regulatory subunits and all functional domains is depicted in Fig.
1a,b. The E545K mutant was built with a single substitution of the glutamic acid E545 to lysine 545K. Subsequent to introducing the mutation and after minimization (see below), the position
of E/K545 backbone nitrogen atom in the helical (p110α) domain remained almost the same allowing the formation of a hydrogen bond with the neighboring L380 backbone oxygen of nSH2 (p85α)
(Fig. 1c). All simulations were performed with the GROMACS 5.0.4 software55, using the AMBER99SB-ILDN all-atom force field56. To decrease the computational cost, the system was solvated in a
rhombic dodecahedron box.The TIP3P potential was used for modeling water molecules57. The number of the water molecules in the mutant simulations was 110982, while in the WT 110996. The
long-range electrostatic interactions were treated using the fast smooth particle-mesh Ewald summation method. The temperature during simulations was kept constant at 310 K using the
Nosé-Hoover thermostat with a time constant of 1 ps. The pressure was isotropically maintained at 1 atm using the Parrinello-Rahman coupling with a time constant of 5 ps and compressibility
of 4.5e-5 bar−1. A time step of 2 fs was used with all bond lengths constrained using the LINCS algorithm. The non-bonded potential energy functions (electrostatic and van der Waals) were
smoothly decaying between cut-off distances 0.8–1.0 nm. Prior to MD simulations, both structures were relaxed by 10,000 steps of energy minimization using the steepest descent algorithm,
followed by positional restraint equilibration first in the NVT and then in the NPT ensemble for 200 ps each. Finally, unbiased MD simulations were carried out with the atomic coordinates of
the systems saved every 2 ps. Two simulations were performed using the WT structure for 1.7 μs in total. In the absence of relevant literature that indicates the protonation state of
crucial PI3Kα residues such as 545K, for our initial simulations, we have used the PROPKA algorithm58,59 to predict residue protonation states. PROPKA predicted that K379, in the
microenvironment of the charged 545K and other positively-charged residues in the vicinity, should be unprotonated, while in the microenvironment of the E545 it should be protonated. We,
thus, performed four independent simulations with the mutant residue K379 unprotonated (total aggregation time 3.6 μs) and four with the mutant residue K379 protonated (total aggregation
time 7.2 μs). The trajectory was analyzed using GROMACS v5.0.455 and VMD51 tools. To assess the intrinsic flexibility and the apparent plasticity of the protein, maps of distance
fluctuations were constructed following the procedure reported by Chiappori _et al_.46. In this work, we also use the dynamical network analysis method, which constructs networks of
biomolecules constituting nodes (atoms) and edges (connecting atoms), weighted by correlation data. This method has been previously applied to study residue networks of several proteins and
protein complexes43,50,52,60,61,62,63,64 and explore putative allosteric communication pathways8. Using this method, time-dependent positional correlations between residues are calculated.
Based on these residue-correlated motions a network representation of the protein is constructed, where the residues (or sets of atoms) are the nodes of the network, which are connected to
each other by links (edges) that depend on the node interaction strength. The method outputs the shortest paths between residues (nodes) as the most dominant mode of their communication.
Edges that most frequently belong in short paths are named _“critical edges”_ and the nodes connected by these edges are established as _“critical nodes”_ for allosteric signal transduction.
The last 200 ns of all reported simulations were used for the dynamical network analysis. All methods are presented in detail in the Supplementary Information. DATA AVAILABILITY All data
(input files, output files, trajectories) have been deposited and can be freely accessed at https://repo.vi-seem.eu/handle/21.15102/VISEEM-254. REFERENCES * Cantley, L. C. The
phosphoinositide 3-kinase pathway. _Science_ 296, 1655–1657, https://doi.org/10.1126/science.296.5573.1655 (2002). Article ADS CAS PubMed Google Scholar * Yuan, T. L. & Cantley, L.
C. PI3K pathway alterations in cancer: variations on a theme. _Oncogene_ 27, 5497–5510, https://doi.org/10.1038/onc.2008.245 (2008). Article CAS PubMed PubMed Central Google Scholar *
Samuels, Y. _et al_. High frequency of mutations of the PIK3CA gene in human cancers. _Science_ 304, 554, https://doi.org/10.1126/science.1096502 (2004). Article CAS PubMed Google Scholar
* Ikenoue, T. _et al_. Functional analysis of PIK3CA gene mutations in human colorectal cancer. _Cancer Res_ 65, 4562–4567, https://doi.org/10.1158/0008-5472.CAN-04-4114 (2005). Article
CAS PubMed Google Scholar * Isakoff, S. J. _et al_. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. _Cancer Res_ 65, 10992–11000,
https://doi.org/10.1158/0008-5472.CAN-05-2612 (2005). Article CAS PubMed Google Scholar * Samuels, Y. _et al_. Mutant PIK3CA promotes cell growth and invasion of human cancer cells.
_Cancer Cell_ 7, 561–573, https://doi.org/10.1016/j.ccr.2005.05.014 (2005). Article CAS PubMed Google Scholar * Rudd, M. L. _et al_. A unique spectrum of somatic PIK3CA (p110alpha)
mutations within primary endometrial carcinomas. _Clin Cancer Res_ 17, 1331–1340, https://doi.org/10.1158/1078-0432.CCR-10-0540 (2011). Article CAS PubMed PubMed Central Google Scholar
* Janku, F. _et al_. PIK3CA mutations in advanced cancers: characteristics and outcomes. _Oncotarget_ 3, 1566–1575, https://doi.org/10.18632/oncotarget.716 (2012). Article PubMed PubMed
Central Google Scholar * Janku, F. _et al_. Assessing PIK3CA and PTEN in early-phase trials with PI3K/AKT/mTOR inhibitors. _Cell Rep_ 6, 377–387,
https://doi.org/10.1016/j.celrep.2013.12.035 (2014). Article CAS PubMed PubMed Central Google Scholar * Gymnopoulos, M., Elsliger, M. A. & Vogt, P. K. Rare cancer-specific mutations
in PIK3CA show gain of function. _Proc Natl Acad Sci USA_ 104, 5569–5574, https://doi.org/10.1073/pnas.0701005104 (2007). Article ADS CAS PubMed PubMed Central Google Scholar * Meyer,
D. S. _et al_. Expression of PIK3CA mutant E545K in the mammary gland induces heterogeneous tumors but is less potent than mutant H1047R. _Oncogenesis_ 2, e74,
https://doi.org/10.1038/oncsis.2013.38 (2013). Article CAS PubMed PubMed Central Google Scholar * Bauer, T. M., Patel, M. R. & Infante, J. R. Targeting PI3 kinase in cancer.
_Pharmacol Ther_ 146, 53–60, https://doi.org/10.1016/j.pharmthera.2014.09.006 (2015). Article CAS PubMed Google Scholar * Bhat-Nakshatri, P. _et al_. Molecular Insights of Pathways
Resulting From Two Common PIK3CA Mutations in Breast Cancer. _Cancer Res_. https://doi.org/10.1158/0008-5472.CAN-15-3174 (2016). Article PubMed Google Scholar * Huang, C. H. _et al_. The
structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. _Science_ 318, 1744–1748, https://doi.org/10.1126/science.1150799 (2007). Article
ADS CAS PubMed Google Scholar * Miller, M. S. _et al_. Structural basis of nSH2 regulation and lipid binding in PI3Kalpha. _Oncotarget_ 5, 5198–5208,
https://doi.org/10.186362/oncotarget.2263 (2014). * Zhao, Y. _et al_. Crystal Structures of PI3Kalpha Complexed with PI103 and Its Derivatives: New Directions for Inhibitors Design. _ACS Med
Chem Lett_ 5, 138–142, https://doi.org/10.1021/ml400378e (2014). Article ADS CAS PubMed Google Scholar * Yang, H. _et al_. Discovery of a Potent Class of PI3Kalpha Inhibitors with
Unique Binding Mode via Encoded Library Technology (ELT). _ACS Med Chem Lett_ 6, 531–536, https://doi.org/10.1021/acsmedchemlett.5b00025 (2015). Article CAS PubMed PubMed Central Google
Scholar * Mandelker, D. _et al_. A frequent kinase domain mutation that changes the interaction between PI3Kalpha and the membrane. _Proc Natl Acad Sci USA_ 106, 16996–17001,
https://doi.org/10.1073/pnas.0908444106 (2009). Article ADS PubMed PubMed Central Google Scholar * Gkeka, P. _et al_. Investigating the structure and dynamics of the PIK3CA wild-type
and H1047R oncogenic mutant. _PLoS Comput Biol_ 10, e1003895, https://doi.org/10.1371/journal.pcbi.1003895 (2014). Article CAS PubMed PubMed Central Google Scholar * Gkeka, P.,
Papafotika, A., Christoforidis, S. & Cournia, Z. Exploring a non-ATP pocket for potential allosteric modulation of PI3Kalpha. _J Phys Chem B_ 119, 1002–1016,
https://doi.org/10.1021/jp506423e (2015). Article CAS PubMed Google Scholar * Zhao, L. & Vogt, P. K. Helical domain and kinase domain mutations in p110alpha of phosphatidylinositol
3-kinase induce gain of function by different mechanisms. _Proc Natl Acad Sci USA_ 105, 2652–2657, https://doi.org/10.1073/pnas.0712169105 (2008). Article ADS PubMed PubMed Central
Google Scholar * Yu, J., Wjasow, C. & Backer, J. M. Regulation of the p85/p110alpha phosphatidylinositol 3′-kinase. Distinct roles for the n-terminal and c-terminal SH2 domains. _J Biol
Chem_ 273, 30199–30203, https://doi.org/10.1074/jbc.273.46.30199 (1998). Article CAS PubMed Google Scholar * Yu, J. _et al_. Regulation of the p85/p110 phosphatidylinositol 3′-kinase:
stabilization and inhibition of the p110alpha catalytic subunit by the p85 regulatory subunit. _Mol Cell Biol_ 18, 1379–1387, https://doi.org/10.1128/MCB.18.3.1379 (1998). Article CAS
PubMed PubMed Central Google Scholar * Nolte, R. T., Eck, M. J., Schlessinger, J., Shoelson, S. E. & Harrison, S. C. Crystal structure of the PI 3-kinase p85 amino-terminal SH2 domain
and its phosphopeptide complexes. _Nat Struct Biol_ 3, 364–374, https://doi.org/10.1038/nsb0496-364 (1996). Article CAS PubMed Google Scholar * Miled, N. _et al_. Mechanism of two
classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. _Science_ 317, 239–242, https://doi.org/10.1126/science.1135394 (2007). Article ADS CAS PubMed Google
Scholar * Lee, J. Y., Engelman, J. A. & Cantley, L. C. Biochemistry. PI3K charges ahead. _Science_ 317, 206–207, https://doi.org/10.1126/science.1146073 (2007). Article CAS PubMed
Google Scholar * Chaussade, C., Cho, K., Mawson, C., Rewcastle, G. W. & Shepherd, P. R. Functional differences between two classes of oncogenic mutation in the PIK3CA gene. _Biochem
Biophys Res Commun_ 381, 577–581, https://doi.org/10.1016/j.bbrc.2009.02.081 (2009). Article CAS PubMed Google Scholar * Hon, W. C., Berndt, A. & Williams, R. L. Regulation of lipid
binding underlies the activation mechanism of class IA PI3-kinases. _Oncogene_ 31, 3655–3666, https://doi.org/10.1038/onc.2011.532 (2012). Article CAS PubMed Google Scholar * Carson, J.
D. _et al_. Effects of oncogenic p110alpha subunit mutations on the lipid kinase activity of phosphoinositide 3-kinase. _Biochem J_ 409, 519–524, https://doi.org/10.1042/BJ20070681 (2008).
Article CAS PubMed Google Scholar * Burke, J. E., Perisic, O., Masson, G. R., Vadas, O. & Williams, R. L. Oncogenic mutations mimic and enhance dynamic events in the natural
activation of phosphoinositide 3-kinase p110alpha (PIK3CA). _Proc Natl Acad Sci USA_ 109, 15259–15264, https://doi.org/10.1073/pnas.1205508109 (2012). Article ADS PubMed PubMed Central
Google Scholar * Marino, K. A., Sutto, L. & Gervasio, F. L. The effect of a widespread cancer-causing mutation on the inactive to active dynamics of the B-Raf kinase. _J Am Chem Soc_
137, 5280–5283, https://doi.org/10.1021/jacs.5b01421 (2015). Article CAS PubMed Google Scholar * Gao, K., He, H., Yang, M. & Yan, H. Molecular dynamics simulations of the Escherichia
coli HPPK apo-enzyme reveal a network of conformational transitions. _Biochemistry_ 54, 6734–6742, https://doi.org/10.1021/acs.biochem.5b01012 (2015). Article CAS PubMed Google Scholar
* Huber, R. G., Fan, H. & Bond, P. J. The Structural Basis for Activation and Inhibition of ZAP-70 Kinase Domain. _PLoS Comput Biol_ 11, e1004560,
https://doi.org/10.1371/journal.pcbi.1004560 (2015). Article ADS CAS PubMed PubMed Central Google Scholar * Liu, H. _et al_. A single residue substitution accounts for the significant
difference in thermostability between two isoforms of human cytosolic creatine kinase. _Sci Rep_ 6, 21191, https://doi.org/10.1038/srep21191 (2016). Article ADS CAS PubMed PubMed Central
Google Scholar * Morando, M. A. _et al_. Conformational Selection and Induced Fit Mechanisms in the Binding of an Anticancer Drug to the c-Src Kinase. _Sci Rep_ 6, 24439,
https://doi.org/10.1038/srep24439 (2016). Article ADS CAS PubMed PubMed Central Google Scholar * Pisani, P., Caporuscio, F., Carlino, L. & Rastelli, G. Molecular Dynamics
Simulations and Classical Multidimensional Scaling Unveil New Metastable States in the Conformational Landscape of CDK2. _PLoS One_ 11, e0154066, https://doi.org/10.1371/journal.pone.0154066
(2016). Article CAS PubMed PubMed Central Google Scholar * Pucheta-Martinez, E. _et al_. An Allosteric Cross-Talk Between the Activation Loop and the ATP Binding Site Regulates the
Activation of Src Kinase. _Sci Rep_ 6, 24235, https://doi.org/10.1038/srep24235 (2016). Article ADS CAS PubMed PubMed Central Google Scholar * Herbert, C. _et al_. Molecular mechanism
of SSR128129E, an extracellularly acting, small-molecule, allosteric inhibitor of FGF receptor signaling. _Cancer Cell_ 23, 489–501, https://doi.org/10.1016/j.ccr.2013.02.018 (2013). Article
CAS PubMed Google Scholar * Echeverria, I., Liu, Y., Gabelli, S. B. & Amzel, L. M. Oncogenic mutations weaken the interactions that stabilize the p110alpha-p85alpha heterodimer in
phosphatidylinositol 3-kinase alpha. _FEBS J_ 282, 3528–3542, https://doi.org/10.1111/febs.13365 (2015). Article CAS PubMed PubMed Central Google Scholar * Kalsi, N., Gopalakrishnan,
C., Rajendran, V. & Purohit, R. Biophysical aspect of phosphatidylinositol 3-kinase and role of oncogenic mutants (E542K & E545K). _J Biomol Struct Dyn_, 1–11,
https://doi.org/10.1080/07391102.2015.1127774 (2016). * Burke, J. E. & Williams, R. L. Dynamic steps in receptor tyrosine kinase mediated activation of class IA phosphoinositide
3-kinases (PI3K) captured by H/D exchange (HDX-MS). _Adv Biol Regul_ 53, 97–110, https://doi.org/10.1016/j.jbior.2012.09.005 (2013). Article CAS PubMed PubMed Central Google Scholar *
Masterson, L. R., Mascioni, A., Traaseth, N. J., Taylor, S. S. & Veglia, G. Allosteric cooperativity in protein kinase A. _Proc Natl Acad Sci USA_ 105, 506–511,
https://doi.org/10.1073/pnas.0709214104 (2008). Article ADS PubMed PubMed Central Google Scholar * McClendon, C. L., Kornev, A. P., Gilson, M. K. & Taylor, S. S. Dynamic
architecture of a protein kinase. _Proc Natl Acad Sci USA_ 111, E4623–4631, https://doi.org/10.1073/pnas.1418402111 (2014). Article ADS CAS PubMed PubMed Central Google Scholar *
Kornev, A. P. & Taylor, S. S. Dynamics-Driven Allostery in Protein Kinases. _Trends Biochem Sci_ 40, 628–647, https://doi.org/10.1016/j.tibs.2015.09.002 (2015). Article CAS PubMed
PubMed Central Google Scholar * Malmstrom, R. D., Kornev, A. P., Taylor, S. S. & Amaro, R. E. Allostery through the computational microscope: cAMP activation of a canonical signalling
domain. _Nat Commun_ 6, 7588, https://doi.org/10.1038/ncomms8588 (2015). Article ADS PubMed Google Scholar * Chiappori, F., Merelli, I., Colombo, G., Milanesi, L. & Morra, G.
Molecular mechanism of allosteric communication in Hsp70 revealed by molecular dynamics simulations. _PLoS Comput Biol_ 8, e1002844, https://doi.org/10.1371/journal.pcbi.1002844 (2012).
Article ADS CAS PubMed PubMed Central Google Scholar * Daily, M. D. & Gray, J. J. Allosteric communication occurs via networks of tertiary and quaternary motions in proteins. _PLoS
Comput Biol_ 5, e1000293, https://doi.org/10.1371/journal.pcbi.1000293 (2009). Article ADS CAS PubMed PubMed Central Google Scholar * Amor, B. R., Schaub, M. T., Yaliraki, S. N. &
Barahona, M. Prediction of allosteric sites and mediating interactions through bond-to-bond propensities. _Nat Commun_ 7, 12477, https://doi.org/10.1038/ncomms12477 (2016). Article ADS
CAS PubMed PubMed Central Google Scholar * Allain, A. _et al_. Allosteric pathway identification through network analysis: from molecular dynamics simulations to interactive 2D and 3D
graphs. _Faraday Discuss_ 169, 303–321, https://doi.org/10.1039/c4fd00024b (2014). Article ADS CAS PubMed Google Scholar * Sethi, A., Eargle, J., Black, A. A. & Luthey-Schulten, Z.
Dynamical networks in tRNA:protein complexes. _Proc Natl Acad Sci USA_ 106, 6620–6625, https://doi.org/10.1073/pnas.0810961106 (2009). Article ADS PubMed PubMed Central Google Scholar *
Humphrey, W. & Schulten, D. A. K VMD: visual molecular dynamics. _J Mol Graph_ 14, 33–38, https://doi.org/10.1016/0263-7855(96)00018-5 (1996). Article CAS PubMed Google Scholar *
Vanwart, A. T., Eargle, J., Luthey-Schulten, Z. & Amaro, R. E. Exploring residue component contributions to dynamical network models of allostery. _J Chem Theory Comput_ 8, 2949–2961,
https://doi.org/10.1021/ct300377a (2012). Article CAS PubMed PubMed Central Google Scholar * Pirola, L. _et al_. Activation loop sequences confer substrate specificity to
phosphoinositide 3-kinase alpha (PI3Kalpha). Functions of lipid kinase-deficient PI3Kalpha in signaling. _J Biol Chem_ 276, 21544–21554, https://doi.org/10.1074/jbc.M011330200 (2001).
Article CAS PubMed Google Scholar * Henzler-Wildman, K. & Kern, D. Dynamic personalities of proteins. _Nature_ 450, 964–972, https://doi.org/10.1038/nature06522 (2007). Article ADS
CAS PubMed Google Scholar * Pronk, S. _et al_. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. _Bioinformatics_ 29, 845–854,
https://doi.org/10.1093/bioinformatics/btt055 (2013). Article CAS PubMed PubMed Central Google Scholar * Aliev, A. E. _et al_. Motional timescale predictions by molecular dynamics
simulations: case study using proline and hydroxyproline sidechain dynamics. _Proteins_ 82, 195–215, https://doi.org/10.1002/prot.24350 (2014). Article CAS PubMed PubMed Central Google
Scholar * Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water _J. Chem. Phys_. 79,
https://doi.org/10.1063/1.445869 (1983). Article ADS CAS Google Scholar * Olsson, M. H. M., Søndergaard, C. R., Rostkowski, M. & Jensen, J. H. PROPKA3: Consistent Treatment of
Internal and Surface Residues in Empirical pKa Predictions. _Journal of Chemical Theory and Computation_ 7, 525–537, https://doi.org/10.1021/ct100578z (2011). Article CAS PubMed Google
Scholar * Søndergaard, C. R., Olsson, M. H. M., Rostkowski, M. & Jensen, J. H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa
Values. _Journal of Chemical Theory and Computation_ 7, 2284–2295, https://doi.org/10.1021/ct200133y (2011). Article CAS PubMed Google Scholar * Scarabelli, G. & Grant, B. J. Mapping
the structural and dynamical features of kinesin motor domains. _PLoS Comput Biol_ 9, e1003329, https://doi.org/10.1371/journal.pcbi.1003329 (2013). Article ADS CAS PubMed PubMed
Central Google Scholar * Guo, C. & Zhou, H.-X. Unidirectional allostery in the regulatory subunit RIα facilitates efficient deactivation of protein kinase A. _Proceedings of the
National Academy of Sciences_ 113, E6776–E6785, https://doi.org/10.1073/pnas.1610142113 (2016). Article CAS Google Scholar * Yao, X.-Q. _et al_. Dynamic Coupling and Allosteric Networks
in the α Subunit of Heterotrimeric G Proteins. _Journal of Biological Chemistry_ 291, 4742–4753, https://doi.org/10.1074/jbc.M115.702605 (2016). Article CAS PubMed Google Scholar *
Musille, P. M., Kossmann, B. R., Kohn, J. A., Ivanov, I. & Ortlund, E. A. Unexpected Allosteric Network Contributes to LRH-1 Co-regulator Selectivity. _Journal of Biological Chemistry_
291, 1411–1426, https://doi.org/10.1074/jbc.M115.662874 (2016). Article CAS PubMed Google Scholar * Miao, Y., Nichols, S. E., Gasper, P. M., Metzger, V. T. & McCammon, J. A.
Activation and dynamic network of the M2 muscarinic receptor. _Proc Natl Acad Sci USA_ 110, 10982–10987, https://doi.org/10.1073/pnas.1309755110 (2013). Article ADS PubMed PubMed Central
Google Scholar Download references ACKNOWLEDGEMENTS The initial part of this work was co-funded by the NSRF2007–2013, the European Regional Development Fund and national resources, under
the grant “Cooperation” [No. 09ΣΥN 11-675]. This work has also been supported by a Marie Curie Reintegration Grant (FP7-PEOPLE-2009-RG, No256533). We acknowledge PRACE for awarding us access
to the computational facility CURIE at GENCI@CEA, France. This work was further supported by computational time granted from the Greek Research & Technology Network (GRNET) in the
National HPC facility - ARIS- under project ID pr001034-PI3Kα-E545K. ZC was co-funded by the European Commission under the H2020 Research Infrastructures Contract No. 675121 (Project
VI-SEEM). We also thank Giorgio Colombo, Paraskevi Gkeka, and Dimitris Dellis for useful discussions. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Biomedical Research Foundation, Academy of
Athens, 4 Soranou Ephessiou, 11527, Athens, Greece Hari Leontiadou, Ioannis Galdadas, Christina Athanasiou & Zoe Cournia Authors * Hari Leontiadou View author publications You can also
search for this author inPubMed Google Scholar * Ioannis Galdadas View author publications You can also search for this author inPubMed Google Scholar * Christina Athanasiou View author
publications You can also search for this author inPubMed Google Scholar * Zoe Cournia View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS H.L.
and I.G. performed simulations, H.L., I.G., C.A. and Z.C. designed research, analysed the data and wrote the paper. All authors reviewed the manuscript. CORRESPONDING AUTHOR Correspondence
to Zoe Cournia. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER'S NOTE: Springer Nature remains neutral with regard
to jurisdictional claims in published maps and institutional affiliations. ELECTRONIC SUPPLEMENTARY MATERIAL SUPPORTING INFORMATION RIGHTS AND PERMISSIONS OPEN ACCESS This article is
licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give
appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in
this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative
Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a
copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Leontiadou, H., Galdadas, I., Athanasiou, C. _et al._
Insights into the mechanism of the _PIK3CA_ E545K activating mutation using MD simulations. _Sci Rep_ 8, 15544 (2018). https://doi.org/10.1038/s41598-018-27044-6 Download citation *
Received: 13 June 2016 * Accepted: 04 May 2018 * Published: 19 October 2018 * DOI: https://doi.org/10.1038/s41598-018-27044-6 SHARE THIS ARTICLE Anyone you share the following link with will
be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt
content-sharing initiative KEYWORDS * Helical Domain * Allosteric Network * Spontaneous Detachment * PI3Kα Activity * N-SH2 Domain