P31–43, an undigested gliadin peptide, mimics and enhances the innate immune response to viruses and interferes with endocytic trafficking: a role in celiac disease
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Celiac disease (CD) is an autoimmune disease characterized by inflammation of the intestinal mucosa due to an immune response to wheat gliadins. Some gliadin peptides are resistant
to intestinal digestion (e.g., A-gliadin P31–43) and induce a stress/innate immune response, but the reason why they are dangerous in the intestines of patients with CD is unknown. In the
present study, P31–43 activated IFN-α, a mediator of the innate immune response in CD, in the intestine of subjects with CD and an enterocyte cell line, CaCo-2. P31–43 cooperated with a
viral ligand to activate the TLR7 pathway by interfering with endocytic trafficking. Based on these results, the vesicular pathway regulates the innate/inflammatory response to viral ligands
and bioactive dietary peptides. Suggesting that together with viral infections, alimentary proteins able to mimic and potentiate the innate immune response to viruses, can trigger an
autoimmune disease such as CD. SIMILAR CONTENT BEING VIEWED BY OTHERS MTOR SUSTAINS INFLAMMATORY RESPONSE IN CELIAC DISEASE Article Open access 01 July 2020 MOLECULAR AND IN VIVO STUDIES OF
A GLUTAMATE-CLASS PROLYL-ENDOPEPTIDASE FOR COELIAC DISEASE THERAPY Article Open access 01 August 2022 COMPLEX REGULATORY EFFECTS OF GUT MICROBIAL SHORT-CHAIN FATTY ACIDS ON IMMUNE TOLERANCE
AND AUTOIMMUNITY Article Open access 01 March 2023 INTRODUCTION Celiac disease (CD) is an autoimmune disease caused by the loss of oral tolerance to gluten, a protein contained in wheat,
barley and rye. The disease is characterized by an enteropathy with inflammatory and structural changes that result in remodelling of the small intestinal mucosa. These changes are the
consequence of mucosal inflammation resulting from a Th1 response to certain gliadin peptides (e.g., the 33-mer A-gliadin peptide) presented by human leucocyte antigen 2 or 8 (HLA-DQ)1 and
activation of innate immune pathways. The activation of these pathways may be mediated by several factors, including other gliadin peptides, e.g., A-gliadin peptide P31–432, not presented by
HLA-DQ2 or 83. Both the 33-mer and 25-mer (P31–55) containing the peptides P57–68 and P31–43, respectively, are very resistant to hydrolysis by gastric, pancreatic and intestinal proteases.
Thus, these peptides are active _in vivo_ in the celiac intestine after gluten ingestion4,5,6. Interleukin 15 (IL15) is a major mediator of the proliferative and innate immune response of
the celiac intestine to gliadin7,8 through cooperation with epidermal growth factor (EGF)7,9,10. The mechanisms by which P31–43 might induce the innate immune response and enterocyte
proliferation have recently been attributed to effects on the endocytic compartment7,11. In both celiac enterocytes and CaCo-2 cells, P31–43 localizes to the early endosomes and delays
vesicular trafficking10,11,12. P31–43, but not P57–68, shares sequence similarity with a region of the growth factor regulated tyrosine kinase substrate (HRS) needed for its correct
endocytic localization. HRS is a key molecule involved in regulating endocytic maturation that is localized on the membranes of early endocytic vesicles12. In CaCo-2 cells, P31–43, but not
P57–68, interferes with the correct localization of HRS to early endosomes, delaying the maturation of the endocytic vesicles12. Consequently, P31–43 induces two important effects: (a) it
delays endocytic maturation and (b) it alters the recycling pathway. A delay in endocytic maturation reduces the degradation of epidermal growth factor receptor (EGFR) and other receptor
tyrosine kinases (RTKs), which are endocytosed by these vesicles. This delay prolongs their activation, resulting in increased proliferation, actin remodelling and other biological effects.
The alteration of the recycling pathway can direct more IL15 receptor alpha (IL15Rα) to the cell surface, enhancing the trans presentation of IL15/IL15Rα in epithelial cells7. Type 1
interferons also play a role in the loss of oral tolerance to gluten in patients with CD. In fact, interferon-alpha (IFN-α) is dysregulated in patients with CD, and IFN-α therapy can induce
CD in some genetically susceptible individuals. In addition, rotavirus infections are associated with an increased incidence of CD13. Moreover, the combination of viral infections and
dietary gliadin causes an enteropathy in normal mice14. The cellular rotavirus receptor is Toll-like receptor 7 (TLR7)15. TLR7 is an endosomal receptor that specifically recognizes the viral
mRNA and is regulated by endosomal trafficking. The signalling pathway initiated by TLR7 when it is engaged by selected viral ligands, including the TLR7-specific ligand loxoribine (LOX),
induces the formation of a myeloid differentiation primary response 88 (MyD88)/TLR7 complex that requires endosomal trafficking to be activated. Subsequently, the activated complex induces
the phosphorylation of mitogen-activated protein kinase (MAPK) and nuclear factor-κB (NF-κB) activation, ultimately increasing the levels of IFN-α and myxovirus resistance protein 1 (MxA),
an antiviral protein that can embed the viral particles16. Interestingly, HRS is also a key factor in endosomal TLR7 and Toll-like receptor 9 (TLR9) trafficking; in fact, it is necessary for
the ubiquitin-dependent targeting of TLR9 to the lysosomes17. Based on these observations, mechanisms regulating vesicular trafficking are central to viral infections response. In the
present study, we investigated whether the A-gliadin peptide P31–43 could mimic and reinforce the IFN-α mediated innate immune response to viruses in biopsies from patients with CD and a
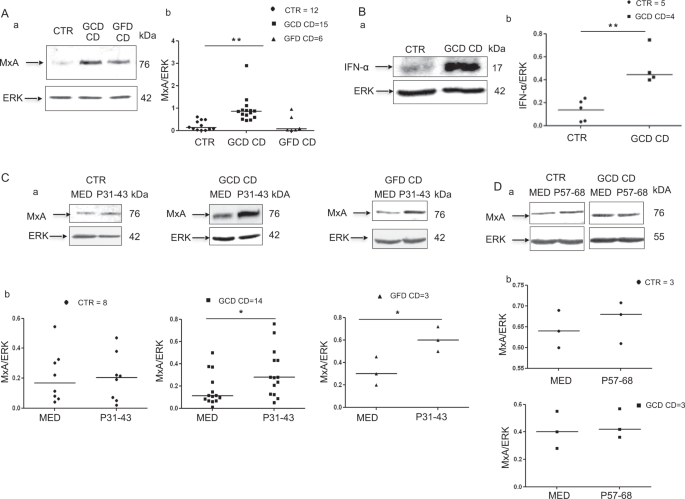
gliadin-responsive intestinal cell line, CaCo-2, by interfering with endocytic trafficking. RESULTS IN SMALL INTESTINAL BIOPSIES, MXA AND IFN-Α ARE EXPRESSED AT HIGHER LEVELS IN PATIENTS
WITH CD ON A GCD AND THE EXPRESSION OF BOTH PROTEINS IS INDUCED BY P31–43 IN PATIENTS WITH CD ON A GLUTEN-CONTAINING DIET (GCD) OR A GLUTEN-FREE DIET (GFD) The IFN-α pathway was activated in
CD biopsies in previous reports13,18. We confirmed these observations by analysing levels of the MxA protein in biopsies from patients with CD on a GCD or GFD. MxA levels were only
increased in patients with CD on a GCD (Fig. 1A). Specifically, the ratio of MxA to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) proteins increased from 0.2 ± 0.2 in the controls to 0.9
± 0.59 (_p_ = 0.006) in patients on a GCD, whereas it was only 0.28 ± 0.39 in the biopsies from patients on a GFD. In addition, levels of the IFN-α protein were increased in patients with CD
on a GCD (Fig. 1B). _In vitro_ P31–43, but not P57–68, treatment increased MxA levels in cultures of the intestinal biopsies from patients with CD on either diet. Values for cultures
obtained from subjects on a GCD (0.3 ± 0.14, _p_ = 0.038) and GFD (0.6 ± 0.10, _p_ = 0.039) were greater than values for cultures of the untreated biopsies (0.16 +/− 0.14 and 0.3 +/− 0.12,
respectively). Levels of the IFN-α 7 and 17 mRNAs were also increased after treatment with P31–43 in biopsies from patients with CD on a GCD (data not shown). In cultured biopsies from the
control subjects (CTR), P31–43 had no effect on the levels of the MxA protein (Fig. 1C) or IFN-α 7 and 17 mRNAs (data not shown). Thus, the IFN-α pathway was activated by the gliadin peptide
P31–43 in the small intestine of patients with CD. SIMILAR TO THE VIRAL LIGAND LOX, P31–43 ACTIVATES THE TLR7 PATHWAY We investigated the effects of the gliadin peptide P31–43 on CaCo-2
cells, an intestinal epithelial cell line that is responsive to gliadin, to obtain a better understanding of the mechanisms by which this peptide activates the IFN-α pathway. We first
investigated the specificity and toxicity of the P31–43 peptide in CaCo-2 cells. The specificity of the effects of P31–43 was confirmed by examining extracellular signal-regulated kinase
(ERK) activation in CaCo-2 cells after treatment with permuted P31–43 (alanine scanning). Most of the mutations reduced ERK phosphorylation (pY-ERK) (Supplemental Fig. 1A). The toxicity of
P31–43 was analysed using Trypan blue staining, a vital colorant. As shown in Supplemental Fig. 1B, treatment with 100 μg/ml P31–43 and P57–68 peptides had no effect on cell viability.
Interestingly, the amount of P31–43 used in our experiments was not substantially different from the amount that is presumably generated in the intestine during the digestion of wheat
flour6. Moreover, a dose response assay using pY-ERK levels as a readout of the effects of P31–43 and P57–68 showed that treatment with 50 and 100 μg/ml P31–43 activated ERK. P57–68 did not
activate ERK at any concentration tested (Supplemental Fig. 1C). We compared the effects of P31–43 and the viral ligand LOX on the activation of the TLR7 pathway in CaCo-2 cells. LOX
specifically activates the MyD88-dependent TLR7 signalling pathway via TLR716. Moreover, CaCo-2 cells in general and, specifically, our clone do not express the other endosomal receptor,
TLR9 (19 and not shown), confirming that the CaCo-2 cell line is a good model with which to study TLR7 activation in epithelial intestinal cells. We compared the effects of P31–43 on the
activation of the TLR7 pathway in CaCo-2 cells with those of LOX, beginning with the formation of the MyD88/TLR7 complex and subsequent activation of mitogen-activated protein kinases
(MAPKs). Figure 2A shows the increase in formation of the MyD88/TLR7 complex induced by the viral ligand LOX and by P31–43 after stimulation for 30 min or 3 h. The amount of TLR7 in the
complex with MyD88 in CaCo-2 cells increased from 0.16 ± 0.13 in the untreated sample to 1.8 ± 0.33 and 2 ± 0.47 in samples treated with LOX for 30 min or 3 h, respectively. Similarly, the
gliadin peptide P31–43 induced a comparable increase in the level of TLR7 in the complex with MyD88 at 30 min (1.5 ± 0.53) and 3 h (1.2 ± 0.49). We next investigated the effects of LOX and
P31–43 on the levels of the TLR7 and MyD88 proteins and found that TLR7 levels were significantly increased in cells treated with LOX for 6 h, (_p_ < 0.01) or P31–43 for 30 min (_p_ <
0.001) or 3 h (_p_ < 0.05). MyD88 levels were also increased at all time points after LOX stimulation. In contrast, after P31–43 treatment, increases in the MyD88 levels were
statistically significant only after 30 min of stimulation (Fig. 2B). The time-dependent differences in the effects of LOX and P31–43 potentially suggest that the mechanisms through which
each stimulus exerts its effects are not the same. The MyD88/TLR7 complexes induced both by LOX and P31–43 could activate downstream signalling by activating MAPKs, ERK, c-Jun N-terminal
kinase (JNK) and protein 38 (p38). Specifically, as shown in Fig. 2C, LOX and P31–43 induced similar increases in the levels of the phosphorylated forms of pY-ERK, JNK (pY-JNK) and p38
(pY-p38) with very similar kinetics. Based on these data, P31–43 activates the same TLR7 pathway in intestinal epithelial cells that is specifically activated by the viral ligand LOX. P31–43
AND THE VIRAL LIGAND LOX INDUCE THE EXPRESSION OF MARKERS OF THE ACTIVATION OF INFLAMMATION AND INNATE IMMUNITY Activation of TLR7 and 9 leads to an innate immune response (increased IFN-α
levels) and an inflammatory response (NF-κB activation)20. LOX and P31–43 treatments increased NF-κB phosphorylation in CaCo-2 cells from 0.45 ± 0.06 in control cells to 0.93 ± 0.1 and 0.86
± 0.08, respectively, as shown in Fig. 3A. We investigated the levels of the MxA protein, a downstream effector of IFN-α activation to follow the activation of the IFN-α pathway. Both LOX
and P31–43 treatments induced a statistically significant increase in levels of the MxA protein at all time points investigated (Fig. 3B). The control gliadin peptide P57–68, which does not
delay vesicular trafficking12, was not effective at promoting the formation of the TLR7/MyD-88 complex, increasing the levels of the TLR7, MyD-88, MxA and IFN-α proteins or activating NF-κB
(Supplemental Fig. 2). The levels of the IFN-α 7 and 17 mRNAs were also analysed after P31–43 and LOX treatments. Levels of the IFN-α 7 and 17 mRNAs were increased to similar extents after
an overnight (ON) treatment with LOX or P31–34 (Fig. 3C). Cooperation between viral ligands and gliadin peptides induces enteropathy in mice14. We investigated whether LOX and P31–43
cooperate to induce the activation of the IFN-α pathway in our system. As shown in Fig. 3D, stimulation of CaCo-2 cells with suboptimal concentrations of LOX or P31–43 alone did not increase
levels of either the MxA protein or IFN-α mRNA. However, when LOX and P31–43 were added together at suboptimal concentrations, levels of both the MxA protein (_p_ < 0.01) and IFN-α mRNA
(_p_ < 0.001) were increased, indicating the synergistic cooperation between the viral ligand and gliadin peptide. THE INCREASE IN MXA LEVELS OBSERVED IN RESPONSE TO P31–43 AND LOX
TREATMENTS DEPENDS ON THE FORMATION OF THE MYD88/TLR7 COMPLEX The engagement of TLR7 by the viral ligand LOX induces downstream signalling pathways that depend on the formation of the
MyD88/TLR7 complex16. We have confirmed this model in the present study, by showing that the increase in the level of the MxA protein observed after LOX treatment was prevented by silencing
of either the MyD88 or TLR7 protein. The P31–43-induced increase in the level of the MxA protein was also prevented by MyD88 or TLR7 silencing, as shown in Fig. 4A and B, indicating that the
activation of the IFN-α pathway depended on the TLR7 activation in response to both LOX and P31–43 treatments. A control scrambled siRNA had no effect on the P31–43- or LOX-induced
increases in levels of the MxA, TLR7 and MyD88 proteins (Fig. 4C). Silencing of TLR7 and MyD88 proteins reduced the levels of the TLR7 and MyD88 proteins. Silencing of TLR7 had no effect on
levels of the MyD88 protein, and silencing of MyD88 had no effect on levels of the TLR7 protein (Fig. 4D). As shown in our previous studies, P31–43 activates EGFR and other receptors by
delaying their intracellular decay through its effect on vesicular trafficking9,10,12. Interestingly, the EGFR receptor complexed with TLR7 after treatment with LOX or P31–43, as shown in
Supplemental Fig. 3. This finding confirms that EGFR transduces signals from several different receptors. P31–43 AND LOX TREATMENTS DELAY TLR7 TRAFFICKING IN THE EARLY ENDOCYTIC COMPARTMENT
Viruses can interfere with vesicular trafficking at various levels during the infection of a cell20. In particular, the MyD88/TLR7 complex that forms in response to the viral ligand LOX
requires endosomal trafficking for activation16. Moreover, P31–43 is known to delay endocytic trafficking from early to late endocytic vesicles10,11,12. We compared the trafficking of TLR7
from early vesicles (early endocytic antigen, EEA1-positive) to late vesicles (lysosome-associated membrane proteins, LAMP 2-positive) after LOX and P31–43 stimulation. TLR7 exhibited
greater co-localization with EEA1 and less co-localization with LAMP2 in samples treated with either LOX and P31–43 than in the untreated sample (Supplemental Fig. 4). We evaluated the
trafficking of TLR7 after P31–43 treatment in time-course experiments. P31–43, but not P57–68, delayed TLR7 trafficking in EEA1-positive vesicles after 3 h of treatment (Supplemental Fig.
5). In conclusion, both the viral ligand and the gliadin peptide P31–43 delayed TLR7 trafficking to the early endocytic compartment. THE DELAY OF THE MATURATION FROM EARLY TO LATE VESICLES
ACTIVATED THE TLR7 PATHWAY P31–43 delayed endocytic maturation by interfering with the correct localization of HRS on endocytic vesicles10,11,12. HRS is a key molecule required for the
maturation of early to late endocytic vesicles21. We tested the hypothesis that the delay in the maturation of early to late vesicles could, _per se_, activate the TLR7 pathway. We tested
this hypothesis by inducing a delay in the maturation of the vesicles through the silencing of the HRS protein (si-HRS)12 and analysed the trafficking of TLR7 to the early and late
compartments. Under these conditions, more TLR7 was co-localized with the EEA1-positive early vesicles (Supplementary Fig. 4) and less with the LAMP2-positive late vesicles, similar to the
effects described after LOX and P31–43 treatments (Supplementary Fig. 4). We next studied the effects of si-HRS on the activation of the TLR7 pathway. As read outs of TLR7 activation, we
analysed the formation of the MyD88/TLR7 complex (Fig. 5A), expression of MxA (Fig. 5B) and IFN-α (Fig. 5C) and the activation of NF-κB (Fig. 5D) after HRS silencing. Similar to the effects
of the gliadin peptide P31–43 and LOX, the amount of TLR7 in complexes with MyD88 was significantly increased compared to the untreated sample (_p_ = 0.03). Levels of the MxA (_p_ = 0.02),
IFN-α 7 (_p_ = 0.0009) and 17 (_p_ = 0.04) mRNAs and the phosphorylated form of NF-κB (p = 0.03) were increased after HRS silencing (Fig. 5). Additionally, levels of both the TLR7 and MyD88
proteins were increased after si-HRS treatment (Fig. 5B). Control si-mRNA had no effect on HRS expression (Fig. 5Bc). In conclusion, the alterations in vesicular trafficking induced by
si-HRS activated the TLR7 pathway and the IFN-α-mediated immune response, as well as NF-κB-mediated inflammation. DISCUSSION In the present study, the undigested A-gliadin peptide P31–43
activated the IFN-α pathway in the intestines of patients with CD and in the gliadin-responsive7,9,10,12,22,23,24,25 enterocyte cell line, CaCo-2. P31–43 could induced the IFN-α-mediated
innate immune response in CaCo-2 cells by activating the TLR7 pathway, similar to the viral ligand LOX. Both the viral ligand LOX and P31–43 delayed the trafficking of TLR7 from the early
endocytic compartment. Moreover, P31–43 and LOX induced increased complex formation and levels of the TLR7 and MyD88 proteins, subsequently activating MAPKs, the innate immune response
(increase in levels of IFN-α and the antiviral protein MxA), and an inflammatory response (increased phosphorylation of NF-κB). Finally, MyD88 and TLR7 silencing prevented complex formation
and downstream pathway activation after either LOX or P31–43 treatment, indicating that complex formation is necessary for both stimuli to exert their effects. Interestingly, LOX and P31–43
cooperated to induce the activation of the IFN-α pathway. Specifically, when LOX and P31–43 were applied together at suboptimal concentrations, levels of both the MxA protein and IFN-α mRNA
were increased. Cooperation between viral ligands and gliadin peptides, including P31–43, has been shown to induce enteropathy in mice14,26. Subsequently, we tested the hypothesis that
P31–43 activated the IFN-α pathway by interfering with endocytic trafficking. This hypothesis is based on two main points. First, P31–43 delayed the maturation of the early to late endocytic
vesicles by interfering with the correct endocytic localization of HRS12. Second, TLR7 is an endosomal receptor that responds to viral ligands by activating the IFN-α pathway, and its
activation is regulated by vesicular trafficking16,17. These findings imply that vesicular trafficking plays a central role in modulating the innate immune activation in the host-pathogen
response. We induced a delay in the maturation of endocytic trafficking by silencing HRS, a master regulator of endocytic maturation12, to confirm the role of the delay in vesicular
trafficking in IFN-α activation. HRS silencing mimicked all the measured effects of P31–43 on the TLR7 pathway: the delay in TLR7 trafficking to the early vesicles, the increases in the
formation of the MyD88/TLR7 complex and protein levels, and the increases in levels of the IFN-α 7/17 mRNAs and MxA protein and NF-κB activation. These data are consistent with the
hypothesis that the mechanism by which P31–43 activates the TLR7 pathway is mediated by a delay in endocytic trafficking. In fact, endocytosis exerts many effects on signalling. The
endocytic pathway and signalling pathways are regulated reciprocally27. The TLR7/MyD88 complex could form in the delayed endocytic vesicles either because the delay in maturation alters the
lipid composition and/or pH of vesicles or by other mechanisms present in cells28. Type 1 interferons play a role in the loss of oral tolerance to gluten in patients with CD13,18. In the
present study, we confirmed that IFN-α is a mediator of the innate immune response in patients with CD by showing an increase in the levels of the IFN-α and MxA proteins and the IFN-α 7 and
17 mRNAs in the intestinal biopsies from patients with CD on a GCD. The activation of the IFN-α pathway may have been induced by dietary gluten. In fact, the gliadin peptide P31–43 activated
the IFN-α pathway in biopsies from patients with CD on a GFD and GCD, but not biopsies from controls, indicating a specific effect of gliadin on the intestines of patients with CD.
Interestingly, we observed a constitutive alteration of the vesicular trafficking in cells from patients with CD that is characterized by a delay in the maturation of early to the late
vesicles, which predisposed the celiac cells to the activation of the innate immune response to gliadin (manuscript submitted). This finding could explain the specific effect of P31–43 on
the activation of the IFN-α pathway in biopsies from patients with CD. Based on the results presented here, viral infections and alimentary proteins, which are able to mimic and potentiate
the innate immune response to viruses, trigger an autoimmune disease. In addition to CD, other autoimmune diseases result from the interactions of several factors, including genetics and the
environment. Type 1 interferon activation is linked to autoimmunity not only in CD but also in several other human diseases, including type 1 diabetes29. Unlike CD, genetic polymorphisms
that directly activate the type 1 interferon pathways are present in some of these diseases, although these alterations alone are not sufficient to induce the disease. The penetrance of
these genetic profiles is determined by mostly unknown environmental factors, including disturbances in the microbiota and/or frequent viral infections29. In the present study, another
environmental factor may be an alimentary protein, such as gliadin. Thus, gluten itself might be a risk factor for these conditions30. METHODS CELL CULTURE, MATERIALS AND TREATMENTS CaCo-2
cells were grown for 5–6 days in Dulbecco’s Modified Eagle’s Medium (DMEM) (GIBCO, San Giuliano Milanese, Italy) supplemented with 10% fetal calf serum (FCS, GIBCO), 100 units/ml of
penicillin-streptomycin (GIBCO), and 1 mM glutamine (GIBCO). The medium was changed every two days. Lipopolysaccharide (LPS)-free synthetic peptides (Inbios, Naples, Italy) (>95% pure,
evaluated by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry) were obtained using Ultrasart-D20 filtration (Sartorius AG, Gottingen, Germany). The level of LPS
in these peptides was below the detection threshold (i.e., <0.20 EU/mg), as assessed using the QCL-1000 kit (Cambrex Corporation, NJ). The sequence of P31–43 was LGQQQPFPPQQPY and that of
P57–68 was QLQPFPQPQLPY. The peptides were used at a concentration of 100 μg/ml10. The guanosine analog/TLR7 ligand loxoribine (7-allyl-7,8 dihydro-8-oxo-guanosine) from InvivoGen (San
Diego, CA, USA) was used at a concentration of 1 mM16. EEA1 AND LAMP STAINING CaCo-2 cells were seeded on glass coverslips for 2 days. Than they were stained for 1 hour at room temperature
with anti-TLR7 and anti EEA1 or LAMP2 antibody after fixation with 3% paraformaldeide for 5 minutes at room temperature and permeabilization with Triton (Biorad, Milan, Italy) 0.2% for 3
minutes at room temperature. Secondary antibodies Alexa-488 conjugated (Invitrogen, Milan, Italy) anti-rabbit for TLR; anti-goat Alexa-546 (Invitrogen, Milan, Italy) for EEA1; anti-mouse
Alexa-546 (Invitrogen, Milan, Italy) for LAMP2 were added to the coverslips for 1 hour at room temperature. Nuclei were stained with Topro-3 iodide conjugate (642/661) (Invitrogen, Milan,
Italy). The coverslips were then mounted on glass slides and observed by confocal microscope (LSM 510 Zeiss). Twenty to 30 cells were observed in each sample. Images were generated with a
confocal microscope LSM 510 Zeiss. Co-localization analysis was performed with AIS Zeiss software. Magnification of the micrographs was the same for all the figures shown (63x objective, 2X
zoom). COLOCALIZATION ANALYSIS Samples were examined with a Zeiss LSM 510 laser scanning confocal microscope. We used Argon/2 (458, 477, 488, 514 nanometers) and HeNe1 (543 nanometers) and
HeNe2 (633 nanometers) excitation lasers, which were switched on separately to reduce cross-talk of the three fluorochromes. The green and the red emissions were separated by a dichroic
splitter (FT 560) and filtered (515-to 540-nm band-pass filter for green and >610-nm long pass filter for red emission). A threshold was applied to the images to exclude about 99% of the
signal found in control images. The weighted co-localization coefficient represents the sum of intensity of co-localizing pixels in channels 1 and 2 as compared to the overall sum of pixel
intensities above threshold. This value could be 0 (no co-localization) or 1 (all pixels co-localize). Bright pixels contribute more than faint pixels. The co-localization coefficient
represents the weighted colocalization coefficients of Ch1 (red) with respect to Ch2 (green) for each experiment25,26. SILENCING EXPERIMENTS Silencing experiments were performed with two
different silencing mRNAs for HRS (Hs HRS 5 and Hs HRS 6), MyD88 (Hs MYD88 2 and Hs MYD88 8), TLR7 (Hs TLR7 6 and Hs TLR7 8) with similar results. Non-specific siRNA (MAPK1) was used for
transfection efficiency (not shown) and scrambled mRNA sequences was used to test specificity of the silencing (All Stars Negative). All silencing mRNAs were all purchased from QIAGEN,
Milan, Italy. Transfections were carried out using the HIPerFect Transfection Reagent following the manufacturer’s instructions (QIAGEN, Milan, Italy). Briefly, CaCo-2 cells were incubated
in standard growth conditions, and 6 μg of siRNA was diluted in 1 ml of culture medium without serum to give a final siRNA concentration of 50 nM. Twenty microliters of HIPerFect
Transfection reagent were added to the siRNA mixture with vortexing, and the transfection mixture was added drop wise onto the cells and Incubated for 72 h. The cells were then processed for
immunoprecipitation and for Western blot (WB) analysis. WESTERN BLOTTING CaCo-2 cells grown as before were stimulated with P31–43 or LOX for various times at 37 °C. The cells were washed
twice and resuspended in lysis buffer (50 mM Tris-HCl, pH 7.4, 1 mM EDTA, 1 mM EGTA, 5 mM MgCl2, 150 mM NaCl, 1% Triton, 1 mM PMSF, 1 mM VO4, 100× Aprotinin, and 50× LAP, all purchased from
Sigma, Milan, Italy, except LAP, which was obtained from Roche, Milan, Italy). The cell lysates were analyzed using SDS-PAGE with a standard running buffer (25 mM Trizma, 192 mM Glycine, and
0.1% SDS) and were transferred onto nitrocellulose membranes (Whatman Gmbh, Dassel, Germany) using transfer buffer (25 mM Trizma, 192 mM Glycine, 0.1% SDS, and 20% methanol, all purchased
from Sigma-Aldrich, Milan, Italy). The membranes were blocked with 5% non-fat dry milk and were probed with goat anti-MxA, rabbit anti-ERK1/2 and mouse anti-pY-ERK1/2, (Santa Cruz, Milan,
Italy), mouse anti-IFN-α (RD System, Milan, Italy), mouse anti-GAPDH, mouse anti-tubulin (Sigma-Aldrich, Milan, Italy), rabbit anti-TLR7, rabbit anti-MyD88, rabbit anti-pY- NF-κB, rabbit
anti-NF-κB, mouse anti-pY-p38, rabbit anti-p38, rabbit anti-pY-JNK and rabbit anti- JNK (Cell Signaling, Euroclene, Milan, Italy) and mouse anti-HRS (Alexix Biochemicals, San Diego, CA,
USA). The bands were visualized using ECL (GE Healthcare, Amersham, Buckinghamshire, UK) with exposure times of 2–10 min. The band intensity was evaluated by integrating all the pixels of
the band after subtraction of the background to calculate the average of the pixels surrounding the band9. IMMUNOPRECIPITATION Lysates were prepared as described previously9, and the protein
concentration was measured using a Bio-Rad protein assay kit (Munchen, Germany). Equal amounts of the cell lysates (2 mg of protein) were used for immunoprecipitation. MyD88 was
immunoprecipitated for 24 h using the rabbit anti-MyD88 polyclonal antibody (Cell Signaling Euroclone, Milan Italy). EGFR was immunoprecipitated for 24 h using the anti-EGFR polyclonal
antibody (Cell Signaling Euroclone, Milan, Italy). Following immunoprecipitation, the complexes were isolated using protein A/G magnetic beads and were washed 3× in lysis buffer. The pellet
was resuspended in 35 μl of 2× Laemmli buffer (Sigma, Milan, Italy). The samples were heated to 95–100 °C for 5 minutes and were micro centrifuged for 1 minute at 14,000 × g. The samples
were loaded (30 μl) onto SDS-PAGE gels (10%). The proteins were transferred to nitrocellulose membranes (Whatman Gmbh, Dassel, Germany), and the blots were probed for TLR7 with anti-TLR7
(Santa Cruz Biotech, Milan, Italy), for MyD88 with mouse anti-MyD88 (Abcam, Milan, Italy) and for EGFR with anti-EGFR polyclonal antibody (Cell Signaling Euroclone, Milan, Italy). PCR
ANALYSIS cDNAs were generated from the total RNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA)9. The resulting cDNA samples were subjected
to cycles of PCR amplification, followed by real-time PCR using TaqMan® preAmp Master Mix Kit protocol (Applied Biosystems, PN 4366127, Foster City, CA, USA). Each TaqMan Gene Expression
assay consisted of two sequence-specific PCR primers and a TaqMan assay-FAM dye-labelled MGB probe. Eighty nanograms of total cDNA was used for each replicate assay. Three replicates were
run for each sample in a 96-well plate format. The endogenous control gene used was beta-2-microglobulin (B2 M). The assays were run with 2× Universal PCR Master Mix without UNG
(uracil-N-glycosylase) using an Applied Biosystems 7300 Real-Time PCR System and universal cycling conditions (10 min at 95 °C; 15 sec at 95 °C, 1 min at 60 °C for 40 cycles). ORGAN CULTURE
STUDIES For organ culture studies, biopsy fragments from the duodenum were obtained from CD patients with villous atrophy on GCD, controls, affected by gastroesophageal reflux, and CD
patients on GFD. The age range for all subjects was 2 to 17 years. GFD patients had negative serology (anti-tTg antibodies between 0 and 1.5 U/ml and EMA negative) and normal biopsy (Marsh
T0–1). GCD-CD patients with villous atrophy (Marsh T3a-c) had a positive serology (anti-tTg antibodies >30 U/ml and EMA positive) (Table 1). Anti-tTg antibodies were measured using the
Eurospital kit, EU-tTG. Biopsy fragments were used to evaluate the expression of the MxA and IFN-α proteins using Western blotting. Intestinal biopsies from the GCD CD patients, the GFD CD
subjects and control subjects were cultured9 with P31–43 (100 μg/ml) or P57–68 (100 μg/ml) or medium alone for 24 h to examine MxA and IFN-α expression. ETHICAL STATEMENTS The protocol for
this study was approved by the Ethical Committee of the University “Federico II”, Naples, Italy (ethical approval: C.E. n. 230/05). Biopsy fragments for organ culture studies were obtained
from the duodenum of untreated patients with active and remission CD and controls affected by gastroesophageal reflux. Written informed consent was obtained from patients or from the next of
kin, caretakers, or guardians on behalf of the minors/children participants involved in our study. The Authors confirm that all methods were performed in accordance with the relevant
guidelines and regulations. STATISTICAL ANALYSES Graphpad prism 5 (Graphpad Software, San Diego, CA, USA) was used for statistical analysis and graphic representation. Statistical analyses
of differences were performed using Student’s _t_-test. A _p_ value < 0.05 was considered statistically significant. One-way Anova, Bonferroni corrected (_p_ value < 0.05 was
considered statistically significant) was used for multiple comparisons as indicated in the figure legends. REFERENCES * Sollid, L. M. Molecular basis of celiac disease. _Ann Rev Immunol_
18, 53–81 (2000). Article CAS Google Scholar * Maiuri, L. _et al_. Association between innate response to gliadin and activation of pathogenic T cells in celiac disease. _Lancet_ 362,
30–37 (2003). Article PubMed CAS Google Scholar * Zimmer, K. P. _et al_. Endocytotic segregation of gliadin peptide 31–49 in enterocytes. _Gut_ 59, 300–1 (2010). Article PubMed CAS
Google Scholar * Shan, L. _et al_. Structural basis for gluten intolerance in celiac sprue. _Science_ 297, 2275–9 (2002). Article ADS PubMed CAS Google Scholar * Comino, I. _et al_.
Monitoring of gluten-free diet compliance in celiac patients by assessment of gliadin 33-mer equivalent epitopes in feces. _Am J Clin Nutr_ 95, 670 (2012). Article PubMed PubMed Central
CAS Google Scholar * Mamone, G. _et al_. Identification of a peptide from alpha-gliadin resistant to digestive enzymes: implications for celiac disease. _J Chromatogr B Analyt Technol
Biomed Life Sci_ 855, 236–41 (2007). Article PubMed CAS Google Scholar * Barone, M. V., Troncone, R. & Auricchio, S. Gliadin peptides as triggers of the proliferative and
stress/innate immune response of the celiac small intestinal mucosa. _Int J Mol Sci_ 15, 20518–537 (2014). Article PubMed PubMed Central CAS Google Scholar * Abadie, V. & Jabri, B.
IL-15: a central regulator of celiac disease immunopathology. _Immunol Rev_ 260, 221–34 (2014). Article PubMed PubMed Central CAS Google Scholar * Nanayakkara, M. _et al_. An undigested
gliadin peptide activates innate immunity and proliferative signaling in enterocytes: the role in celiac disease. _Am J Clin Nutr_ 98, 1123–35 (2013). Article PubMed CAS Google Scholar
* Barone, M. V. _et al_. Growth factor-like activity of gliadin, an alimentary protein: implications for celiac disease. _Gut_ 56, 480–8 (2007). Article PubMed PubMed Central CAS Google
Scholar * Barone, M. V. & Zimmer, K. P. Endocytosis and transcytosis of gliadin peptides. _Mol Cell pediatr._ 3, 8 (2016). Article PubMed PubMed Central Google Scholar * Barone, M.
V. _et al_. Gliadin peptide P31-43 localises to endocytic vesicles and interferes with their maturation. _Plos One_ 5, e12246 (2010). Article ADS PubMed PubMed Central CAS Google
Scholar * Sangman, M. K., Toufic, M. & Jabri, B. Innate immunity: actuating the gears of celiac disease pathogenesis. _Best pract Res Clin Gastroenterol_ 29, 425–35 (2015). Google
Scholar * Araya, R. E., Jury, J., Bondar, C., Verdu, E. F. & Chirdo, F. G. Intraluminal administration of poly I:C causes an enteropathy that is exacerbated by administration of oral
dietary antigen. _Plos One_ 9, e99236 (2014). Article ADS PubMed PubMed Central CAS Google Scholar * Pane, J. A., Webster, N. L. & Coulson, B. S. Rotavirus activates lymphocytes
from non-obese diabetic mice by triggering toll-like receptor 7 signaling and interferon production in plasmacytoid dendritic cells. _Plos Pathog_ 10, e1003998 (2014). Article PubMed
PubMed Central Google Scholar * Heil, F. _et al_. The Toll-like receptor 7 (TLR7)-specific stimulus loxoribine uncovers a strong relationship within the TLR7, 8 and 9 subfamily. _Eur J
Immunol_ 33, 2987–97 (2003). Article PubMed CAS Google Scholar * Chiang, C. Y. _et al_. Cofactors required for TLR7-and TLR9 dependent innate immune responses. _Cell Host Microbe_ 11,
306–18 (2012). Article PubMed PubMed Central CAS Google Scholar * Monteleone, G. _et al_. Role of interferon alpha in promoting T helper cell type 1 responses in the small intestine in
coeliac disease. _Gut_ 48, 425–9 (2001). Article PubMed PubMed Central CAS Google Scholar * Pedersen, G., Andresen, L., Matthiessen, M. W., Rask-Madsen, J. & Brynskov, J. Expression
of Toll-like receptor 9 and response to bacterial CpG oligodeoxynucleotides in human intestinal epithelium. _Clin Exp Immunol._ 141, 298–306 (2005). Article PubMed PubMed Central CAS
Google Scholar * Lee, B. L. & Barton, G. M. Trafficking of endosomal Toll-like receptors. _Trends Cell Biol_ 24, 360–9 (2014). Article PubMed PubMed Central CAS Google Scholar *
Raiborg, C. & Stenmark, H. Hrs and endocytic sorting of ubiquitinated membrane proteins. _Cell Struct Funct._ 27, 403–8 (2002). Article PubMed CAS Google Scholar * Iacomino, G. _et
al_. Structural analysis and Caco-2 cell permeability of the celiac-toxic A-gliadin peptide 31–55. _J Agric Food Chem._ 61, 1088–96 (2013). Article PubMed CAS Google Scholar * Caputo, I.
_et al_. Gliadin peptides induce tissue transglutaminase activation and ER-stress through Ca2+ mobilization in Caco-2 cells. _Plos One._ 7(9), e45209 (2012). Article ADS PubMed PubMed
Central CAS Google Scholar * Reinke, Y., Zimmer, K. P. & Naim, H. Y. Toxic peptides in Frazer’s fraction interact with the actin cytoskeleton and affect the targeting and function of
intestinal proteins. _Exp Cell Res._ 315, 3442–52 (2009). Article PubMed CAS Google Scholar * Zimmermann, C. _et al_. Epithelial transport of immunogenic and toxic gliadin peptides _in
vitro_. _Plos One._ 9, e113932 (2014). Article ADS PubMed PubMed Central CAS Google Scholar * Araya, R. E. _et al_. Mechanisms of innate immune activation by gluten peptide p31-43 in
mice. _Am J physiol Gastrointest Liver physiol._ 1(311), G40–9 (2016). Article ADS Google Scholar * Scita, G. & Di Fiore, P. P. The endocytic matrix. _Nature._ 28(463), 464–73 (2010).
Article ADS CAS Google Scholar * Fang, C., Wei, X. & Wei, Y. Mitochondrial DNA in the regulation of innate immune responses. _Protein Cell._ 7, 11–6 (2016). Article PubMed CAS
Google Scholar * Todd, J. A. Constitutive antiviral immunity at the expense of autoimmunity. _Immunity_ 40, 164–169 (2014). Article CAS Google Scholar * Antvorskov, J. C. _et al_.
Gluten-Free Diet Only during Pregnancy Efficiently Prevents Diabetes in NOD Mouse Offspring. _J. Diabetes Res_., 3047574 (2016). Download references ACKNOWLEDGEMENTS (1) AIC (Associazione
Italiana Celiachia) project N. 053_FC_2013; (2) POR (progetto Operativo Regionale) Campania 2007/2013. Title of the project: Benessere dalle Biotecnologie: nuovi processi e prodotti per la
Nutraceutica, la Cosmetoceutica e la Nutrizione Umana. (BenTen); (3) Campania Bioscience: Progetto 2 “Progettazione, sviluppo e Produzione di cibi funzionali e/o arricchiti” PON (Progetto
Operativo Nazionale) 2007/2013. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Translational Medical Science (Section of Paediatrics) and ELFID (European Laboratory for the
Investigation of Food-Induced Disease), University of Naples, Federico II, Naples, 80131, Italy Merlin Nanayakkara, Giuliana Lania, Mariantonia Maglio, Renata Auricchio, Cristiana De Musis,
Valentina Discepolo, Erasmo Miele, Riccardo Troncone, Salvatore Auricchio & Maria Vittoria Barone * Department of Medicine, University of Chicago, Chicago, Illinois, 60637, USA Bana
Jabri Authors * Merlin Nanayakkara View author publications You can also search for this author inPubMed Google Scholar * Giuliana Lania View author publications You can also search for this
author inPubMed Google Scholar * Mariantonia Maglio View author publications You can also search for this author inPubMed Google Scholar * Renata Auricchio View author publications You can
also search for this author inPubMed Google Scholar * Cristiana De Musis View author publications You can also search for this author inPubMed Google Scholar * Valentina Discepolo View
author publications You can also search for this author inPubMed Google Scholar * Erasmo Miele View author publications You can also search for this author inPubMed Google Scholar * Bana
Jabri View author publications You can also search for this author inPubMed Google Scholar * Riccardo Troncone View author publications You can also search for this author inPubMed Google
Scholar * Salvatore Auricchio View author publications You can also search for this author inPubMed Google Scholar * Maria Vittoria Barone View author publications You can also search for
this author inPubMed Google Scholar CONTRIBUTIONS Merlin Nanayakkara and Giuliana Lania conducted the experiments, analysed the data and prepared most of the figures. Mariantonia Maglio
cultured the biopsies. Valentina Discepolo analysed the data. Cristiana de Musis conducted the experiments. Renata Auricchio was responsible for the selection and follow-up of the patients.
Erasmo Miele performed the biopsies. Bana Jabri critically revised the manuscript for important intellectual content. Riccardo Troncone obtained the funding and revised the manuscript.
Salvatore Auricchio wrote the paper and analysed and interpreted the data. M. Vittoria Barone designed the study, analysed the data and wrote the paper. CORRESPONDING AUTHOR Correspondence
to Maria Vittoria Barone. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER'S NOTE: Springer Nature remains neutral
with regard to jurisdictional claims in published maps and institutional affiliations. ELECTRONIC SUPPLEMENTARY MATERIAL SUPPLEMENTAL INFORMATION RIGHTS AND PERMISSIONS OPEN ACCESS This
article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as
you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party
material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s
Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Nanayakkara, M., Lania, G., Maglio, M. _et
al._ P31–43, an undigested gliadin peptide, mimics and enhances the innate immune response to viruses and interferes with endocytic trafficking: a role in celiac disease. _Sci Rep_ 8, 10821
(2018). https://doi.org/10.1038/s41598-018-28830-y Download citation * Received: 13 November 2017 * Accepted: 02 July 2018 * Published: 17 July 2018 * DOI:
https://doi.org/10.1038/s41598-018-28830-y SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative