Iron deficiency disrupts embryonic haematopoiesis but not the endothelial to haematopoietic transition
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

In this study, we aimed to explore how cellular iron status affects embryonic haematopoiesis. For this purpose, we used a model of mouse embryonic stem cell differentiation into embryonic
haematopoietic progenitors. We modulated the iron status by adding either the iron chelator Deferoxamine (DFO) for iron deficiency, or ferric ammonium citrate for iron excess, and followed
the emergence of developing haematopoietic progenitors. Interestingly, we found that iron deficiency did not block the endothelial to haematopoietic transition, the first step of
haematopoiesis. However, it did reduce the proliferation, survival and clonogenic capacity of haematopoietic progenitors. Surprisingly, iron deficiency affected erythro-myeloid progenitors
significantly more than the primitive erythroid ones. Erythro-myeloid progenitors expressed less transferrin-receptor on the cell surface and had less labile iron compared to primitive
erythroid progenitors, which could reduce their capacity to compete for scarce iron and survive iron deficiency. In conclusion, we show that iron deficiency could disturb haematopoiesis at
an early embryonic stage by compromising more severely the survival, proliferation and differentiation of definitive haematopoietic progenitors compared to restricted erythroid progenitors.
Embryonic haematopoiesis is an essential and complex process, which supplies blood to the developing embryo and the adult. It generates a wide range of haematopoietic progenitors and stem
cells (HPSCs), from the primitive non-self-renewing erythroid progenitors of the yolk sac to the long-term self-renewing haematopoietic stem cells, which will reside in the adult bone marrow
and continuously generate all blood lineages1,2. Despite its overall complexity, embryonic haematopoiesis can be simplified into a sequence of steps common for all types of HPSCs. The first
step, the endothelial-to-hematopoietic transition (EHT), initiates blood development whereby endothelial cells of a particular type called haemogenic endothelium undergo significant
morphological and transcriptomic changes to become HPSCs3,4,5. Afterwards, the HPSCs proliferate, differentiate, and migrate to colonize the foetal liver and bone marrow2,6,7,8. Recent work
on human9 and mouse embryonic stem cells10,11 as well as on reprogrammed mouse embryonic fibroblasts12 contributed significantly to our knowledge of how transcription factors and growth
factors control embryonic haematopoiesis. Yet the role of iron in the process of embryonic haematopoiesis is not completely understood.
Iron is an essential micronutrient required for catalysis, DNA synthesis, redox reactions and oxygen transport13. Iron deficiency through a knockout of iron import proteins like transferrin
receptor (Tfrc) or Dmt1 (Slc11a2) causes anaemia and embryonic or early postnatal lethality in mice14,15. Nutritional iron deficiency in pregnant females increases the risk of iron
deficiency and iron deficiency anaemia in the offspring, according to animal models and human epidemiological studies16,17. Hypotransferrinaemic hpx/hpx mice18 or mice chimeric for Tfrc
knockout19 have a defect in T lymphoid differentiation, suggesting that the effects of iron deficiency might not be restricted only to the erythroid lineage. We therefore hypothesized that
iron was important for an early step in embryonic haematopoiesis, which is common for all developing blood cells. Thus, we investigated the impact of iron on the process of EHT and early HPC
populations.
To dissect the step of embryonic haematopoiesis when iron is most required, we needed an experimental model of embryonic haematopoiesis where we could change the cellular iron status quickly
and reversibly and measure the effect in real time. To fulfil these requirements, we chose the experimental model of mouse embryonic stem cells progressively differentiating into blood
cells through a haemangioblast stage3, similarly to what happens in yolk sac haematopoiesis3. The haemangioblast stage cultures start as Flk1+ mesoderm and differentiate into a mixed culture
of endothelial, haematopoietic progenitor, and vascular smooth muscle cells3. In these cultures, we modified the cellular iron status by adding either an iron chelator (DFO) to cause iron
deficiency or adding ferric ammonium citrate to induce a state of iron excess20.
In this work, we demonstrated that iron deficiency by DFO did not block EHT itself, but it differentially affected the proliferation, survival and differentiation of early haematopoietic
progenitors. In contrast, iron excess had no adverse effects on haematopoietic progenitors. Thus, our findings offer broader understanding of how iron deficiency could affect embryonic
haematopoiesis16.
We first tested whether iron deficiency would block the first step of embryonic haematopoiesis, which is the endothelial-to-haematopoietic transition (EHT)3,4,5. This hypothesis was based on
previously published evidence that iron chelators were inhibiting the epithelial-to-mesenchymal transition (EMT), a mechanism similar to EHT21.
For this purpose, we differentiated mouse embryonic stem cells22 into hemangioblast cultures3,10,11 and followed the formation of haematopoietic progenitors from hemangioblast as a function
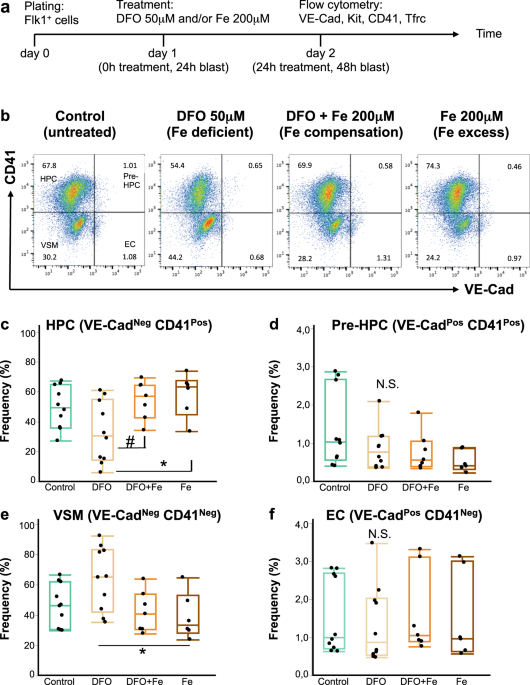
of iron status and time. The time course of our experiments is schematically represented in Fig. 1a. Our hemangioblast cultures were composed of four major cell types as demonstrated in Fig.
1b: vascular smooth muscle cells (VSM), endothelial cells (EC), haematopoietic progenitor cells (HPCs) and cells in an intermediate EHT stage, referred to as Pre-HPCs. These four cell types
were distinguished in flow cytometry by expression of an endothelial marker VE-Cadherin (VE-Cad) and an early haematopoietic marker CD41. Cells expressing neither VE-Cad nor CD41 were
vascular smooth muscle cells; endothelial cells, VE-Cad+ CD41−; HPCs, VE-Cad− CD41+ and Pre-HPCs, VE-Cad+ CD41+.
Iron deficiency does not block endothelial to haematopoietic transition. (a) A schematic timeline of an experiment in blast culture. Flk1+ cells were plated in blast mix on day 0, treated 24
hours later and profiled by flow cytometry for expression of endothelial and hematopoietic markers after another 24 hours. The treatments were: nothing (control), DFO 50 μM (iron-deficient
conditions), DFO + ferric ammonium citrate 200 μM (simultaneous neutralization of iron deficiency, or iron compensation), Fe 200 μM (ferric ammonium citrate for iron excess conditions). (b)
A flow cytometry plot showing expression of CD41 against VE-Cad in day 2 blast (24 hours of treatment). According to the expression of these markers, each blast culture can be divided into 4
quadrants: VE-Cad− CD41− = vascular smooth muscle cells (VSM); VE-Cad+ CD41− = endothelial cells (EC); VE-Cad+ CD41+ = pre-hematopoietic progenitor cells (Pre-HPC); VE-Cad− CD41+ =
hematopoietic progenitor cells (HPC). The experimental conditions are shown from left to right. The Tukey’s boxplots of the different cell type frequencies from flow cytometry experiments
are shown for HPC (c), Pre-HPC (d), VSM (e) and EC (f). Box whiskers show minimum and maximum, the line inside boxes shows median. For control and DFO groups n = 10; for DFO + Fe and Fe
groups n = 6. All groups were analysed by one-way ANOVA followed by Tukey’s multiple comparisons test. For (c) #p = 0.049 and *p = 0.027, for (e) *p = 0.032, for (d,f) N.S. = not
significant.
If iron plays an essential role in EHT, we would expect that the addition of an iron chelator to our hemangioblast cultures would reduce HPCs frequency and cause accumulation of endothelial
cells and/or Pre-HPCs7. DFO addition at 50 μM for 24 hours reduced the frequency and the absolute cell number of HPCs in culture (Fig. 1b,c, Supplementary Fig. 1). However, neither the
frequency nor the cell number of endothelial and Pre-HPCs were increased by DFO (Fig. 1b,d,f, Supplementary Fig. 1), suggesting no accumulation of these cells took place. We further observed
a slight increase in the frequency of vascular smooth muscle cells after DFO treatment (Fig. 1e), but their absolute cell number was not increased (Supplementary Fig. 1).
Excess iron added as 200 μM ferric ammonium citrate together with DFO abrogated all effects of DFO in culture, demonstrating that the observed effects were truly due to iron deficiency. Iron
addition together with DFO or alone did not significantly reduce the frequencies and absolute cell numbers of any cell type compared to untreated control (Fig. 1, Supplementary Fig. 1),
suggesting that in our conditions the given iron concentration was not toxic to cells. The 200 μM of iron should already be in excess to saturate the total transferrin in our culture medium,
since we estimate the total concentration of transferrin in our culture medium as ~10 μM. Our estimation is based on average 140 μM total iron binding capacity of bovine serum23 (equivalent
to ~70 μM transferrin24) plus contribution from human holotransferrin and conditioned medium in our culture.
All haematopoietic progenitors in our cultures can be divided into two major subtypes: definitive KitPos HPCs (Kit+ CD41+) and primitive KitNeg HPCs (Kit- CD41+). The KitPos HPCs mostly
yield multilineage haematopoietic colonies when plated on methylcellulose with appropriate growth factors and are capable of reconstituting irradiated mice albeit transiently, while the
KitNeg HPCs yield mostly primitive erythroid colonies on methylcellulose and have no reconstitution capacity3,25. When we examined the effect of DFO on both kinds of HPCs, we saw that the
frequency of KitPos HPCs was significantly (p