Variable protein homeostasis in housekeeping and non-housekeeping pathways under mycotoxins stress
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Transcript levels are the primary factor determining protein levels, but for the majority of genes, fold changes in transcript levels are larger than the corresponding changes in
protein levels, a phenomenon that is termed “protein homeostasis”. However, this phenomenon is not well characterized in the context of environmental changes. In this study, we sequenced the
entire transcriptome and proteome of chicken primary hepatocytes administered three mycotoxin treatments Aflatoxin B1 (AFB1), Ochoratoxin A (OTA) and Zearalenone (ZEN). Each mycotoxin
induced unique set of differential expressed transcripts and proteins, suggesting variable cytotoxicity and biochemical action in cell. We found a weak positive correlation between
transcript and protein changes, and the transcript changes were higher than the protein changes. Furthermore, we observed pathway-specific protein homeostasis pattern under mycotoxin stress.
Specifically, the “Metabolism”, “Transcription” and “Translation” pathways (housekeeping pathways) showed lower fold changes in protein/mRNA levels than non-housekeeping pathways such as
“Cell growth and death” and “Immune system”. Protein molecular weight had a weak negative effect on protein production, and this effect was stronger for non-housekeeping pathways. Overall,
we hypothesize housekeeping pathways maintain stable protein production for baseline cellular function, whereas non-housekeeping pathways is associated with the fitness response to
environmental stress. SIMILAR CONTENT BEING VIEWED BY OTHERS THE ACUTE TRANSCRIPTIONAL RESPONSES TO DIETARY METHIONINE RESTRICTION ARE TRIGGERED BY INHIBITION OF TERNARY COMPLEX FORMATION
AND LINKED TO ERK1/2, MTOR, AND ATF4 Article Open access 12 February 2021 IDENTIFICATION OF FUNCTIONAL FEATURES UNDERLYING HEAT STRESS RESPONSE IN SPRAGUE–DAWLEY RATS USING MIXED LINEAR
MODELS Article Open access 10 May 2022 ORGANELLE SPECIFIC FLUORESCENT PHENOMICS AND TRANSCRIPTOMIC PROFILING TO EVALUATE CELLULAR RESPONSE TO TRIS(1,3 DICHLORO 2 PROPYL)PHOSPHATE Article
Open access 18 March 2022 INTRODUCTION The central dogma that the information flow in a biological system is generally from DNA to RNA to protein is the cornerstone of modern molecular
biology. Although this flow appears to be straightforward, there is a very complicated relationship between mRNAs and its encoded proteins. Besides of the regulatory networks in
co-/post-transcription processes, protein quantity in cells is influenced by many aspects, including codon composition, ribosomal entry sites, translation rates, protein half-life, mRNA and
protein degeneration rate, protein modulation, folding and transport rates1. With next-generation sequencing breakthroughs in recent years, increasing amounts of large-scale transcriptome
and proteome data are now available, and researchers must disentangle the general rules governing protein production. Although the concept is still controversial2,3, several studies have
characterized a significant positive correlation between mRNA and protein levels (rho > 0.5) in yeast, Drosophila, _Caenorhabditis elegans_, zebrafish, human and plants cells in the
“steady” states4,5,6,7. Thus, mRNA levels are suggested to be the primary factor determining protein levels. Based on these calculations, mRNA levels possibly accounts for 40% to 80% of
protein abundance variance; other factors, such as translation rate, play a relatively minor role in protein production8,9. However, recent observations from fission yeast, human tumors and
evolutionary studies between humans and chimpanzees, revealed that fold changes in mRNA levels are generally larger than fold changes in protein levels10,11,12. This phenomenon is termed as
protein homeostasis that protein production is maintained at a stable level in the cell despite fluctuations in mRNA1. Two concepts reveal the complicated nature of the protein production
process: the first stresses the importance of mRNA quantity in determining protein production, whereas the second indicates that other regulatory mechanisms are also important in determining
a stable protein concentration. Thus, further studies, especially in different species or under different cellular conditions such as environmental pressures, are needed to elucidate
protein homeostasis. Mycotoxins are a group of low-molecular-weight secondary metabolites produced by filamentous fungi13. Approximately 400 mycotoxins have been identified and a dozen have
been recognized as important threats to humans and animals, including aflatoxin, citrinin, ergot alkaloids, fumonisin B1, ochoratoxin A (OTA), patulin, trichothecenes, zearalenone (ZEN) and
so on14. Mycotoxin contamination in crops is widespread around the world, especially in developing countries due to poor preharvest practice and postharvest storage and transportation15. It
is estimated that 25% of world’s crop may be contaminated by mycotoxins. The US Council for Agricultural Science and Technology estimated that the cost of crop losses from aflatoxins,
fumonisins and deoxynivalnol is $932 million USD per year and mitigation cost is $466 million per year16. Some mycotoxins have stable molecular properties which are difficult to be removed
by common practices such as heating or filtering, and as the consequence, consumptions of mycotoxin-containing foods and feeds lead to various pathologic reactions and can even increase
mortality rates13,17,18. Aflatoxin B1 (AFB1), OTA and ZEN are some of the most widely spread mycotoxins in the world. Previous studies reported these mycotoxins have similar toxic actions in
cells including prohibiting RNA and protein synthesize, DNA damage and ROS13,18,19, but none of them quantified the expression profile at the omics level. In this study, we sequenced the
entire transcriptome and proteome of chicken (_Gallus gallus_) primary hepatocytes under AFB1, OTA and ZEN treatments. As in steady-state cells, the protein homeostasis pattern was preserved
under mycotoxin pressure. However, genes from different pathways showed differing levels of protein homeostasis, indicating protein homeostasis was primarily maintained by housekeeping
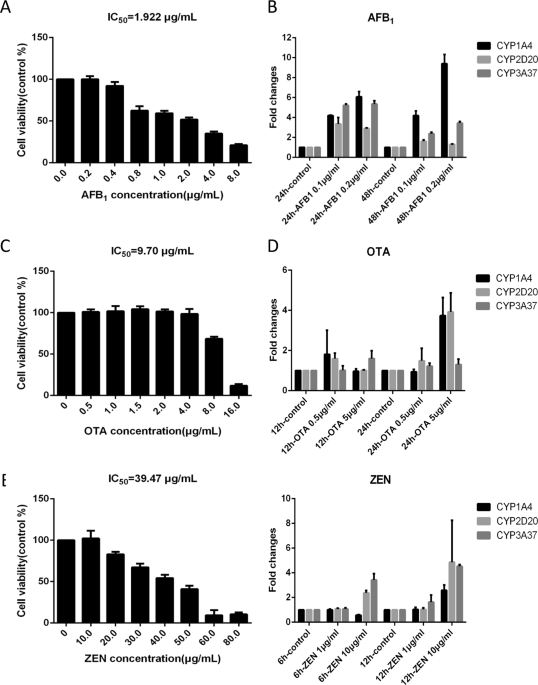
pathways in the cell. RESULTS MYCOTOXINS TREATMENT OF CHICKEN PRIMARY HEPATOCYTES Chicken primary hepatocytes were administrated by three mycotoxins AFB1, OTA and ZEN. We used an MTT assay
to assess cell viability under mycotoxin administration. The mycotoxin concentration was lower than 0.5 μg/mL, 5 μg/mL and 20 μg/mL (AFB1, OTA and ZEN) to maintain cell viability >90%
(Fig. 1). Then, a gradient test was conducted to quantify CYP1A, CYP2D and CYP3A gene expression under different mycotoxin concentration and duration (Fig. 1). We used the lowest and
shortest toxin treatment that induced the expression of all three CYP450s, which were 0.1 μg/mL 24 h for AFB1, 5 μg/mL 24 h for OTA and 10 μg/mL 12 h for ZEN, to administrate the chicken
primary hepatocytes. HIGH-THROUGHPUT SEQUENCING Three biological replicates were collected for transcriptome and proteome sequencing from both untreated hepatocytes as the control, and
mycotoxin-treated hepatocytes. To analyze the transcriptome, ~50 million reads (150-bp paired-end reads generated by Illumina, 6.34 to 7.52 Gb in total) were obtained for each replicate.
High-quality scores (Q20 > 95%), low error rates (0.02%) and stable GC content indicated a high-quality transcript dataset (Table S1). To verify the data consistency among biological
replicates, we applied Pearson’s correlation analysis between all samples (Fig. S1). The correlations were all >0.98, higher than the best practice guideline by ENCODE Consortium
(0.92–0.98). For the proteome analysis, we used iTRAQ (isobaric tag for relative and absolute quantification) technology to quantify relative protein levels between three mycotoxin-treated
and control samples. Spectra from tandem mass spectrometry were searched using MASCOT engine 2.2 (Matrix Science, London UK) against the UniProt chicken database for peptide identification
(https://www.ebi.ac.uk/GOA/chicken_release, Uniprot_Chicken_24083_20150713.fasta). Peptides were quantified by Proteome Discoverer 2.0 (Thermo Fisher Scientific, San Jose, CA) and the false
discovery rate (FDR) was calculated based on a Decoy database search with a cutoff of 0.01. The majority of peptide lengths ranged from 5 to 25 amino acids (Fig. S2), similar to the range
reported previously20. Protein ratios were calculated based on the combined median ratio of unique peptides, and experimental bias was controlled by normalizing the median protein ratio to
1. The final protein ratio was the mean of the three biological replicates (Table S2). DIFFERENTIAL EXPRESSED TRANSCRIPTS AND PROTEINS The set of differentially expressed transcripts (DETs)
were first characterized. We got quantitative expression level for 19,948 transcripts in total, and the DETs were identified based on negative binomial distribution with Benjamini and
Hochberg procedure adjusted p value < 0.0521. Compared to the control treatment, mycotoxin treatment led to intensive transcriptomic changes (Figs 2 and S3). For AFB1, we found 6,994 DETs
(35.1% of total transcripts), including 3,583 up-regulated and 3,411 down-regulated transcripts. For OTA, there were 8,657 DETs (43.4% of total transcripts), including 4,496 up-regulated
and 4,161 down-regulated genes. For ZEN, there were 3,548 DETs (17.8% of total transcripts), including 1,692 up-regulated and 1,856 down-regulated genes (Table S3; Fig. 3). The number of
shared up-regulated and down-regulated transcripts among all three mycotoxins was much lower than the mycotoxin-specific DETs, especially for up-regulated transcripts (Fig. 2A,B). We further
grouped the up-regulated and down-regulated transcripts based on the KEGG pathway definition (Tables 1 and 2). For AFB1 treated samples, the up-regulated transcripts were mainly enriched in
the “Environmental information processing” pathway, including “Cytokine-cytokine receptor interaction”, “ECM-receptor interaction”, “Jak-STAT signaling” and “Cell adhesion molecules (CAMs)”
pathways. For ZEN treated samples, the up-regulated transcripts were mainly enriched in “Genetic information processing” pathway, including “DNA replication”, “Spliceosome”, “RNA
transport”, “Mismatch repair”, “Homologous recombination”, “Base excision repair” and “RNA polymerase” pathways. The down-regulated transcripts were enriched in “Metabolism” and “Cellular
processes” pathways for AFB1, “Ribosome” and “Focal adhesion” pathways for OTA and “Valine, leucine and isoleucine degradation” and “PPAR signaling” pathways for ZEN. We identified much less
number of differentially expressed proteins (DEPs): only 656 (10.3%), 548 (8.6%) and 216 (3.4%) DEPs were between AFB1-, OTA- and ZEN- treated and control samples (Table S3; Figs 2 and S4).
Similar to the transcript dataset, the number of shared DEPs was much lower than the mycotoxin-specific DEPs (Fig. 2). For AFB1 treated samples, the up-regulated proteins were enriched in
“Metabolism” pathway, including “Fructose and mannose metabolism”, “Retinol metabolism” and “Biosynthesis of amino acids” pathways, and cancer related “Chemical carcinogenesis” pathway. For
ZEN, the up-regulated proteins were enriched in “Biosysthesis of amino acids” pathway, and for OTA in “Complement and coagulation cascades” pathway (Table 1). For AFB1 treated samples, the
down-regulated proteins were enriched in “Environmental information processing” pathway, including “ECM-receptor interaction”, “Cell adhesion molecules” and “Cytokine-cytokine receptor
interaction”, and pathways from cellular community and cancer. For OTA treated samples, the down-regulated proteins were enriched in “ECM-receptor interaction” pathway, and for ZEN in
“Insulin signaling pathway”. Although we found some similarities between different mycotoxin administrations, such as “ECM-receptor interaction” pathways were enriched with down-regulated
proteins for both AFB1 and OTA, the overall distribution of DETs and DEPs was quite different for the three mycotoxins. Furthermore, the set of DETs were not consistent with the set of DEPs
(Tables 1 and 2). For AFB1, seven pathways were enriched with up-regulated transcripts but none of them was enriched with up-regulated proteins. Similarly, four pathways were enriched with
up-regulated proteins but none of them was enriched with up-regulated transcripts. This pattern suggests that each pathway may assume a specific mRNA and protein production dynamics under
mycotoxin pressure. PROTEIN HOMEOSTASIS UNDER MYCOTOXIN STRESS We extracted 4,110 genes with quantitative transcript (FPKM > 1) and protein expression values for all three mycotoxins, and
visualized the expression fold changes (Fig. 3). We found a weak significant positive correlation between the transcript and protein changes for all mycotoxins (p < 2.2e-16), and the
correlations for AFB1 (0.29) and ZEN (0.23) were lower compared to previous reports for cells in the “steady” states (rho > 0.5). The transcript level changes were much higher than
protein level changes for most genes, thus the protein homeostasis pattern was preserved under mycotoxin stress (Fig. 2). We further plotted transcript and protein changes for each KEGG
pathway for AFB1, OTA and ZEN (Figs S5–S7). We found certain pathways, such as “Carbohydrate metabolism” and “Nucleotide metabolism” under OTA, showed lower protein /mRNA changes than the
“Signal transduction” and “Cell growth and death” pathways (Fig. 4). More specifically, we used slope (the fitted line for linear regression) to represent the overall protein/mRNA fold
changes. A higher slope indicated higher protein-to-mRNA changes and relaxed constraint on protein homeostasis. We listed the complete results in Table S4 and summarized the results in Table
3. To be conservative, we only showed pathways with correlations >0.3 and p values < 0.01. As most housekeeping genes are associated with the “Metabolism”, “Transcription” and
“Translation” pathways (KEGG 1.1 to 2.2) and most genes in other pathways (KEGG 2.3 to 5.3) are non-housekeeping genes22, we classified the two sets of pathways as housekeeping pathways and
non-housekeeping pathways. In OTA-treated samples, housekeeping pathways showed lower protein/mRNA changes than non-housekeeping pathways (p = 0.001, two-tailed t-test). In AFB1-treated
samples, the overall pattern was the same (p = 0.044). In ZEN-treated samples, we did not observe higher changes in non-housekeeping than housekeeping pathways. The slopes for
non-housekeeping pathways were still higher than the slopes of most housekeeping pathways, but the pattern was distorted by the relatively high slope for “Energy metabolism” (0.293).
Combining the results obtained with all three mycotoxins, we identified several pathways with high protein changes, including “Cell growth and death” (>0.2 for all three mycotoxin),
“Folding, sorting and degradation”, “Replication and repair”, “Membrane transport”, and “Immune system” (>0.2 for two mycotoxin). Overall, we found variable protein homeostasis pattern
under mycotoxin administration. It is tempting to hypothesize a fitness optimization process in cell that the strong constraint on housekeeping pathways is a baseline requirement for the
maintenance of cellular function, and the relaxed constraint on non-housekeeping pathways is primarily related to the functional response to specific mycotoxin pressures. OTHER FACTORS
ASSOCIATED WITH PROTEIN HOMEOSTASIS Molecular weight may be involved in protein homeostasis. It has been hypothesized that protein production cost is primarily determined by protein
molecular weight, and thus, the overproduction of a low-cost protein should have a small fitness effect, whereas the production of a high-cost protein is more tightly controlled23. Thus, the
homeostasis of proteins with high molecular weights should be stronger than the homeostasis of low-molecular-weight proteins. To test this assumption, we calculated the correlation between
molecular weight and protein level changes in the three mycotoxin-treated samples, and we found very weak but significant negative correlations for all three samples (rho −0.029, −0.054 and
−0.23, p < 0.05; Table 4, Fig. 5). This result suggests that molecular weight may only play a minor role or be relevant for a subset of proteins. We further conducted the analyses on
housekeeping and non-housekeeping pathways separately, and found a negative correlation for each analysis (Table 4). The correlation was higher for non-housekeeping pathways than for
housekeeping pathways under AFB1 and OTA treatment. For ZEN treatment, the correlations and p values were similar between the two sets. This result suggests that molecular weight affects
both gene sets, but the effect is stronger for non-housekeeping pathways, consistent with the hypothesis of a relaxed protein homeostasis constraint on these gene sets. Protein location may
also be involved with protein homeostasis. We used MetazSecKB24 to predict chicken protein locations and evaluate the impact of cellular location on protein changes (Tables S5 and S6; Figs
S8–S10). In general, we found that proteins associated with organelle membranes showed higher protein changes than secreted, cell plasma membrane-associated and in-lumen proteins (p = 0.02,
two-tailed t-test). In particular, “Golgi apparatus membrane” and “Nuclear membrane” genes had high slopes (>0.2), and genes in the “Cytoplasm” location had low slope values. The data
suggests that subcellular location is also associated with protein homeostasis under mycotoxin stress. However, gene function and location are closely confounding factors. For example,
membrane proteins primarily function as signal transduction receptors, substrate transporters and enzymes25, and the majority of metabolism and information processing genes are located in
the cytoplasm. Thus, it is necessary to take into account of the two features together and interpret the results with caution. LONG NON-CODING TRANSCRIPT ISOFORM IS NOT THE MECHANISM FOR
PROTEIN HOMEOSTASIS Previous studies reported that the length of 5′ UTRs influenced mRNA translation efficiency; the isoform of long undecoded transcript had poor translational rate compared
to the canonical transcripts26. We analyzed the expression of long UTRs and normal UTRs in the mycotoxin-treated and control samples. The long UTRs were defined as UTRmycotoxin – UTRcontrol
> 500 bp, and normal UTRs were defined as −5 < UTRmycotoxin – UTRcontrol < 5. Only UTRcontrol < 600 bp were included in the analysis27. We did not find association between the
UTR lengths and protein expression levels in our dataset (Fig. S11). Thus, UTR length variation is not the mechanism for protein homeostasis in chicken hepatocytes. DISCUSSION In this study,
we used widespread mycotoxins AFB1, OTA and ZEN to administrate chicken embryo hepatocytes and characterize the transcriptomic and proteomic changes. The high-throughput data indicated that
the three mycotoxins induced variable sets of differential expressed transcripts and proteins, but the overall pattern of protein homeostasis was still preserved. This study systematically
evaluated the association between protein homeostasis and molecular factors, including gene function, protein location and molecular weight, and hypothesis a fitness optimization process in
cell. AFB1 is one the most studied and characterized mycotoxin to date due to the outbreak of turkey X disease in the early 1960s28. It is well established that active metabolites of AFB1
can bind to the N7 position of guanines, and the AFB1-DNA adducts result in GC to TA transversions13. Other biochemical actions include decreasing RNA content and RNA polymerase, suppressing
protein synthesis, interrupting carbohydrate metabolism by decreasing glycogen level and inhibiting electron transport in mitochondria29. Similarly, OTA also has multiple biochemical
actions in cell, including oxidative stress, protein synthesis inhibition and activation or deactivation of specific cell signaling pathways30. ZEN and its derivatives can competitively bind
to the estrogen receptor, leading to various estrogenic syndromes13. Study on cultured Vero cells reveals that ZEN and its derivatives also inhibit protein and DNA synthesis, cell
viability, oxidative damages and over-expression of stress proteins31. Thus, all three mycotoxins have multiple biochemical and molecular actions in cell, but it is unclear whether they
affect the same set of genes/pathways or not. In this study, we found that each mycotoxin had quite specific impact on functional pathways, led to different molecular action and cellular
responses in chicken hepatocytes. The phenomenon are unlikely due to different stress levels posed by these mycotoxins. We controlled the mycotoxin stress on the minimum level to maintain
>90% cell viability throughout the study. The lowest level of mycotoxin concentration and shortest duration time that induced the expression of CYP450s have been chosen to administrate
the chicken cells. Thus, stress levels should not be a major factor leading different cellular responses. Previous studies reported that the iTRAQ technique underestimated the fold change
signals by compressing the isotopic intensity ratios and the underestimation became more obvious for proteins with high changes32,33. Another study estimated the iTRAQ measurement had
~40–50% compression to the true value34. In our study, the changes of protein expression was several fold lower than the transcript expression, thus the technique compression can contribute
to the fold differences, but should not be a major explanation for the protein homeostasis pattern we observed. Another potential problem is that protein production is slower than mRNA
production, and thus, the protein homeostasis we observed may be attributable to a production delay rather than a true stasis effect. However, previous studies showed that protein production
was generally only delayed for 5 or 6 h after the mRNA oscillations9,35. In this study, we applied AFB1 and OTA treatment for 24 h and ZEN for 12 h, which are much longer times than the
reported delay in protein production. Thus, the effects of “production delay” should be minimal in our study. Protein production is much more expensive than mRNA production. Warner _et al_.
estimated that cells divert 50% of their energy to protein production, whereas only 5% of energy is spent on mRNA production36. It costs 2 GTP and 1 ATP for each peptide bond formed, whereas
only 1 NTP is required for nucleotide elongation. Researchers estimated that the total number of proteins is ~1,850- to ~10,000- fold higher than mRNA copy numbers in fission yeast and
mammals10,37,38. Thus, protein production is very costly in term of cellular energy and resources but is crucial for maintaining cellular function. According to evolutionary theory, protein
production should be maximized or constrained according to the fitness of a cell or organism, which is termed as an optimization process39,40. This theory has been tested for a few
phenotypes and genes, such as the production of Lac protein in _Escherichia coli_, but has never been tested at the whole-transcriptome/proteome level. The pathway-specific protein
homeostasis observed in this study fit this evolutionary optimization process and demonstrated selective advantages for cell survival under mycotoxin pressure. Genes involved in specific
functions such as “Cell growth and death” or “Immune system” had relaxed constraints on protein production, demonstrating clear advantages for active responses to the changing environment.
However, the housekeeping pathways experienced a stronger constraint to maintain much more stable protein concentrations for baseline cellular functions. By maintaining stable
concentrations, disruptive signals from toxic molecules do not interfere with core gene production, which in turn reduces toxic cellular effects in response to relatively minor damage.
Homeostasis theories can be soundly described and explained by this cost-benefit optimization theory, but the molecular mechanism underlying buffering remains elusive. Researchers have tried
to explain the pattern from the perspective of translational rates but have found conflicting results. Some evidence suggests that translation rates contribute significantly to protein
buffering41,42, whereas others studies have indicated that translation rates are not important43,44. Dephoure _et al_. reported that translation rate variation was not important for the
large-scale regulation of protein production; however, the protein degradation rate may be a major factor shaping protein homeostasis patterns45. A recent proteomic study in mouse cardiac
cells also supported this hypothesis by revealing that functionally associated protein were co-regulated in degradation process46. Others studies alternatively proposed that the microRNAs
may contribute significantly to mRNA and protein correlations47,48. A new hypothesis suggested that variation of mRNA length altered the meiotic protein expressions levels26. We measured the
expression profile for transcripts with long and normal UTRs and failed to find any association in the chicken model (Fig. S7). But as only a small portion of long non-coding RNAs were
identified in our study due to the sequencing depth limitation, we cannot totally rule out this possibility. Overall, it is unclear what is the main mechanism to buffer the protein variation
and whether different organisms have same mechanism or not, thus more studies are needed in the future to address these issues. METHODS EXPERIMENTAL DESIGN AND STATISTICAL RATIONALE In this
study, we performed transcriptome and proteome sequencing for three mycotoxin-treated samples (AFB1, OTA and ZEN) and one control sample. For each sample, we collected and sequenced three
biological replicates. To detect differentially expressed genes, Benjamini and Hochberg’s approach was used to control the FDR. Pearson’s correlation was used for correlation analysis
because the dataset has a normal distribution. We included all data points in the statistical analyses. CHICKEN PRIMARY HEPATOCYTES ISOLATION AND MYCOTOXIN PREPARATION Eighteen-day
embryonated specific-pathogen-free (SPF) Arbor Acres Broiler chicken eggs were purchased from the Institute of Animal Science, Guangdong Academy of Agricultural Sciences (Guangzhou, China).
All related experiments and methods were performed in accordance with the recommended guidelines and regulations by the Administration of Affairs Concerning Experimental Animals of Guangdong
Province, China. This research was approved (approval number 2015-D010) by Laboratory Animal Ethics Committee of South China Agricultural University. Hepatocytes were prepared using a
collagenase digestion method49. Chicken embryo primary hepatocytes were re-suspended in Williams E medium (Sigma-Aldrich, Shanghai) containing 10% FBS, 100 U/mL penicillin/streptomycin, 10
nM insulin, and 10 nM dexamethasone (Sigma-Aldrich). We added the medium to each culture plate and cultured cells at 37 °C in a humidified incubator with 5.0% CO2. After 5 h, the medium was
removed from adherent cells, and the cells were maintained in Medium 199 (Life Technologies, Shanghai) supplemented with 10% FBS, 100 U/mL penicillin/streptomycin, 10 μg/mL insulin, 1 μg/mL
dexamethasone, and 2 mM L-glutamate (Invitrogen). AFB1, ZEN and OTA were purchased from Pribolab (Qindao, China). All mycotoxins were dissolved in Dimethyl sulfoxide (DMSO). Williams E
medium, Medium 199 and FBS were purchased from Invitrogen (Carlsbad, CA, USA). MTT CELL VIABILITY ASSAY AND QUANTITATIVE RT-PCR Cell viability was assessed according to a previously
described method50. Chicken embryo primary hepatocytes were seeded at a density of 10000 cells/well in 96-well plates and cultured overnight. The hepatocytes were treated with different
concentrations of AFB1 (0–10 μg/mL), ZEN (0–120 μg/mL), or OTA(0–10 μg/mL). After 48 h, 0.5 mg/mL MTT (Sigma-Aldrich, Shanghai) was added to each well. We extracted the total RNA from
mycotoxin-treated or untreated chicken primary hepatocytes using Trizol reagent (Invitrogen, Carlsbad, CA, USA). Total RNA (2 μg) was reverse transcribed using M-MLV reverse transcriptase
(Promega, Madison, WI, USA) with oligo d(T) and random primers (Takara Biotechnology, Dalian, China). The obtained cDNA products were used as templates for CYP1A4, CYP2D20 and CYP3A37
transcript quantification by RT-PCR. GAPDH was used as an internal reference to normalize gene expression. The expression levels were calculated using the 2−ΔΔCt method51. LIBRARY
PREPARATION AND TRANSCRIPTOME SEQUENCING Total RNA purity, concentration and integrity were assessed using NanoPhotometer® spectrophotometry (IMPLEN, CA, USA), Qubit® RNA Assay Kit in a
Qubit® 2.0 Fluorometer (Life Technologies, CA, USA), and RNA Nano 6000 Assay Kit on a Bioanalyzer 2100 system (Agilent Technologies, CA, USA). A total of 3 μg of RNA was extracted per
sample. Sequencing libraries were constructed using NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NEB, USA), then sequenced on an Illumina Hiseq X Ten platform, with paired-end reads
length of 150 bp. TRANSCRIPTOME DATA ANALYSIS Raw data were processed through Perl scripts by removing reads that contained adapter, ploy-N and low quality reads. The Q20, Q30 and GC
contents for each sample were calculated. All downstream analyses were based on the processed clean data. The reference chicken genome and annotation files were downloaded from NCBI with the
GenBank Assembly ID GCA_000002315.3. The index of the reference genome was built using Bowtie v2.2.352, and paired-end clean reads were aligned to the reference genome using TopHat
v2.0.1253. The read numbers mapping to each gene were calculated by HTSeq v0.6.154, and the FPKM of each gene was calculated based on the length of the gene and the reads count mapped to
that gene. Differential expression analysis was performed using the DESeq R package (1.18.0)21, and the statistical enrichment of the differential expression genes in KEGG
(http://www.genome.jp/kegg/) pathways was calculated by KOBAS55,56. SAMPLE PREPARATION, ITRAQ LABELING AND MASS SPECTROMETRY SDT buffer was used for the sample preparation as in the
universal proteomic sample preparation protocol20. For SDS-PAGE separation, 20 µg of proteins was mixed with 5X loading buffer and boiled for 5 min. The proteins were separated on a 12.5%
SDS-PAGE gel (constant current 14 mA, 90 min) and visualized using Coomassie Blue R-250 staining. The peptide mixture of each sample (100 μg) was labeled with iTRAQ reagent according to the
manufacturer’s instructions (Applied Biosystems, Shanghai). The labeled peptides were fractionated by SCX chromatography (GE Healthcare). LC-MS/MS analysis was performed with a Q Exactive
mass spectrometer (Thermo Scientific, Shanghai) and Easy nLC (Proxeon Biosystems, Danmark). MS data was collected based on the most abundant precursor ions from the survey scan (300–1800
m/z). PROTEOME DATA ANALYSIS MS/MS spectra were analyzed using MASCOT engine (Matrix Science, London, UK; version 2.2) with following parameter settings. The search was MS/MS ion search. The
protease used to generate peptides was trypsin. Two missed cleavages were permitted. The mass values were monoisotopic. The list of all fixed modifications considered included
carbamidomethyl (C), and iTRAQ4PLEX (N-terminal), iTRAQ4plex (K). The list of all variable modifications included oxidation (M) and iTRAQ4plex (Y). The peptide mass tolerance for precursor
ions was ±20 ppm. The mass tolerance for fragment ions was 0.1 Da. We used the Decoy database embedded in Proteome Discoverer 2.0 (Thermo Fisher Scientific) to calculate the FDR and set the
cutoff to <0.01 for a peptide. The retrieved sequences were locally searched against the UniProt GOA database (chicken) (https://www.ebi.ac.uk/GOA/chicken_release,
Uniprot_Chicken_24083_20150713.fasta) using NCBI BLAST+ client software (ncbi-blast-2.2.28 + -win32.exe)57 for annotation. Protein ratios were calculated as the median of only the unique
peptides for a protein. To exclude experimental bias, we normalized all peptide ratios using the median protein ratio. Protein quantification was calculated using the normalized spectral
index (SIN)58. Technical repeats were accessed by performing a multivariate Pearson correlation analysis with R. To categorize genes into specific KEGG pathways, we blasted the annotated
proteins against the online KEGG database55. Mapping and ID conversion were facilitated using R package ‘org.Gg.eg.db’ in Bioconductor. Downstream analysis and graphic plotting for
transcriptomic and proteomic data were primarily performed using the Rstudio platform59. DATA AVAILABILITY The proteomic raw data were deposited in the ProteomeXchange Consortium via the
PRIDE partner repository under the dataset identifier PXD008961. The sequenced RNAseq raw data, processed read counts and FPKM file were deposited in GEO database of NCBI (Accession Number
GES112862). REFERENCES * Liu, Y., Beyer, A. & Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. _Cell_ 165, 535–50 (2016). Article CAS Google Scholar *
Fortelny, N., Overall, C. M., Pavlidis, P. & Freue, G. V. C. Can we predict protein from mRNA levels? _Nature_ 547, E19–E20 (2017). Article CAS Google Scholar * Wilhelm, M. _et al_.
Mass-spectrometry-based draft of the human proteome. _Nature_ 509, 582–7 (2014). Article CAS ADS Google Scholar * Ponnala, L., Wang, Y., Sun, Q. & van Wijk, K. J. Correlation of mRNA
and protein abundance in the developing maize leaf. _Plant J_ 78, 424–40 (2014). Article CAS Google Scholar * Beyer, A., Hollunder, J., Nasheuer, H. P. & Wilhelm, T.
Post-transcriptional expression regulation in the yeast Saccharomyces cerevisiae on a genomic scale. _Mol Cell Proteomics_ 3, 1083–92 (2004). Article CAS Google Scholar * Shaik, A. _et
al_. _Functional Mapping of the Zebrafish Early Embryo Proteome and Transcriptome_ (2014). * Lundberg, E. _et al_. Defining the transcriptome and proteome in three functionally different
human cell lines. _Mol Syst Biol_ 6, 450 (2010). Article Google Scholar * Li, J. J., Bickel, P. J. & Biggin, M. D. System wide analyses have underestimated protein abundances and the
importance of transcription in mammals. _PeerJ_ 2, e270 (2014). Article Google Scholar * Jovanovic, M. _et al_. Immunogenetics. Dynamic profiling of the protein life cycle in response to
pathogens. _Science_ 347, 1259038 (2015). Article Google Scholar * Marguerat, S. _et al_. Quantitative analysis of fission yeast transcriptomes and proteomes in proliferating and quiescent
cells. _Cell_ 151, 671–83 (2012). Article CAS Google Scholar * Geiger, T., Cox, J. & Mann, M. Proteomic changes resulting from gene copy number variations in cancer cells. _PLoS
Genet_ 6, e1001090 (2010). Article Google Scholar * Khan, Z. _et al_. Primate transcript and protein expression levels evolve under compensatory selection pressures. _Science_ 342, 1100–4
(2013). Article CAS ADS Google Scholar * Bennett, J. W. & Klich, M. Mycotoxins. _Clin Microbiol Rev_ 16, 497–516 (2003). Article CAS Google Scholar * Cole, R. J. & Cox, R. H.
_Handbook of toxic fungal metabolites_, xvii, 937 p. (Academic Press, New York, 1981). * Peraica, M., Radic, B., Lucic, A. & Pavlovic, M. Toxic effects of mycotoxins in humans. _Bull
World Health Organ_ 77, 754–66 (1999). CAS PubMed PubMed Central Google Scholar * Report, C. Mycotoxins: risks in plant, animal, and human systems. (eds Richard, J. L. & Payne, G.
A.) (Council for Agricultural Science and Technology Task Force Report 2003; No. 139, Ames, Iowa, USA, 2003). * Newberne, P. M. & Butler, W. H. Acute and chronic effects of aflatoxin on
the liver of domestic and laboratory animals: a review. _Cancer Res_ 29, 236–50 (1969). CAS PubMed Google Scholar * Hussein, H. S. & Brasel, J. M. Toxicity, metabolism, and impact of
mycotoxins on humans and animals. _Toxicology_ 167, 101–34 (2001). Article CAS Google Scholar * Butler, W. H. & Neal, G. E. Mode of action and human health aspects of aflatoxin
carcinogenesis. _Ann Nutr Aliment_ 31, 949–56 (1977). CAS PubMed Google Scholar * Wisniewski, J. R., Zougman, A., Nagaraj, N. & Mann, M. Universal sample preparation method for
proteome analysis. _Nat Methods_ 6, 359–62 (2009). Article CAS Google Scholar * Anders, S. & Huber, W. Differential expression analysis for sequence count data. _Genome Biol_ 11, R106
(2010). Article CAS Google Scholar * Eisenberg, E. & Levanon, E. Y. Human housekeeping genes, revisited. _Trends Genet_ 29, 569–74 (2013). Article CAS Google Scholar * Wessely, F.
_et al_. Optimal regulatory strategies for metabolic pathways in Escherichia coli depending on protein costs. _Mol Syst Biol_ 7, 515 (2011). Article Google Scholar * Meinken, J., Walker,
G., Cooper, C. R. & Min, X. J. MetazSecKB: the human and animal secretome and subcellular proteome knowledgebase. _Database (Oxford)_ 2015 (2015). * Almen, M. S., Nordstrom, K. J.,
Fredriksson, R. & Schioth, H. B. Mapping the human membrane proteome: a majority of the human membrane proteins can be classified according to function and evolutionary origin. _BMC
Biol_ 7, 50 (2009). Article Google Scholar * Cheng, Z. _et al_. Pervasive, Coordinated Protein-Level Changes Driven by Transcript Isoform Switching during Meiosis. _Cell_ 172, 910–923 e16
(2018). Article CAS Google Scholar * Rao, Y. S., Wang, Z. F., Chai, X. W., Nie, Q. H. & Zhang, X. Q. Relationship between 5′ UTR length and gene expression pattern in chicken.
_Genetica_ 141, 311–8 (2013). Article CAS Google Scholar * Blout, W. P. Turkey “X” disease. _Turkeys_ 6, 55–77 (1961). Google Scholar * Kiessling, K. H. Biochemical mechanism of action
of mycotoxins. In _Pure and Applied Chemistry_ Vol. 58 327 (1986). * Marin-Kuan, M., Cavin, C., Delatour, T. & Schilter, B. Ochratoxin A carcinogenicity involves a complex network of
epigenetic mechanisms. _Toxicon_ 52, 195–202 (2008). Article CAS Google Scholar * Othmen, Z. O.-B., Golli, E. E., Abid-Essefi, S. & Bacha, H. Cytotoxicity effects induced by
Zearalenone metabolites, α Zearalenol and β Zearalenol, on cultured Vero cells. _Toxicology_ 252, 72–77 (2008). Article CAS Google Scholar * Karp, N. A. _et al_. Addressing accuracy and
precision issues in iTRAQ quantitation. _Mol Cell Proteomics_ 9, 1885–97 (2010). Article CAS Google Scholar * Ow, S. Y., Salim, M., Noirel, J., Evans, C. & Wright, P. C. Minimising
iTRAQ ratio compression through understanding LC-MS elution dependence and high-resolution HILIC fractionation. _Proteomics_ 11, 2341–6 (2011). Article CAS Google Scholar * Ow, S. Y. _et
al_. Balancing robust quantification and identification for iTRAQ: application of UHR-ToF MS. _Proteomics_ 10, 2205–13 (2010). Article CAS Google Scholar * Robles, M. S., Cox, J. &
Mann, M. _In-vivo_ quantitative proteomics reveals a key contribution of post-transcriptional mechanisms to the circadian regulation of liver metabolism. _PLoS Genet_ 10, e1004047 (2014).
Article Google Scholar * Warner, J. R. The economics of ribosome biosynthesis in yeast. _Trends Biochem Sci_ 24, 437–40 (1999). Article CAS Google Scholar * Schwanhausser, B. _et al_.
Global quantification of mammalian gene expression control. _Nature_ 473, 337–42 (2011). Article ADS Google Scholar * Azimifar, S. B., Nagaraj, N., Cox, J. & Mann, M.
Cell-type-resolved quantitative proteomics of murine liver. _Cell Metab_ 20, 1076–87 (2014). Article CAS Google Scholar * Dekel, E. & Alon, U. Optimality and evolutionary tuning of
the expression level of a protein. _Nature_ 436, 588–92 (2005). Article CAS ADS Google Scholar * Orr, H. A. The genetic theory of adaptation: a brief history. _Nat Rev Genet_ 6, 119–27
(2005). Article CAS Google Scholar * Artieri, C. G. & Fraser, H. B. Evolution at two levels of gene expression in yeast. _Genome Res_ 24, 411–21 (2014). Article CAS Google Scholar
* Wang, Z. _et al_. Evolution of gene regulation during transcription and translation. _Genome Biol Evol_ 7, 1155–67 (2015). Article CAS Google Scholar * Battle, A. _et al_. Genomic
variation. Impact of regulatory variation from RNA to protein. _Science_ 347, 664–7 (2015). Article CAS ADS Google Scholar * Albert, F. W., Muzzey, D., Weissman, J. S. & Kruglyak, L.
Genetic influences on translation in yeast. _PLoS Genet_ 10, e1004692 (2014). Article Google Scholar * Dephoure, N. _et al_. Quantitative proteomic analysis reveals posttranslational
responses to aneuploidy in yeast. _Elife_ 3, e03023 (2014). Article Google Scholar * Lau, E. _et al_. Integrated omics dissection of proteome dynamics during cardiac remodeling. _Nat
Commun_ 9, 120 (2018). Article ADS Google Scholar * Ebert, M. S. & Sharp, P. A. Roles for microRNAs in conferring robustness to biological processes. _Cell_ 149, 515–24 (2012).
Article CAS Google Scholar * Herranz, H. & Cohen, S. M. MicroRNAs and gene regulatory networks: managing the impact of noise in biological systems. _Genes Dev_ 24, 1339–44 (2010).
Article CAS Google Scholar * Hamer, M. J. & Dickson, A. J. Control of glycolysis in cultured chick embryo hepatocytes. Fructose 2,6-bisphosphate content and phosphofructokinase-1
activity are stimulated by insulin and epidermal growth factor. _Biochem J_ 269, 685–90 (1990). Article CAS Google Scholar * Mosmann, T. Rapid colorimetric assay for cellular growth and
survival: application to proliferation and cytotoxicity assays. _J Immunol Methods_ 65, 55–63 (1983). Article CAS Google Scholar * Livak, K. J. & Schmittgen, T. D. Analysis of
relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. _Methods_ 25, 402–8 (2001). Article CAS Google Scholar * Langmead, B. & Salzberg,
S. L. Fast gapped-read alignment with Bowtie 2. _Nat Methods_ 9, 357–9 (2012). Article CAS Google Scholar * Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice
junctions with RNA-Seq. _Bioinformatics_ 25, 1105–11 (2009). Article CAS Google Scholar * Anders, S., Pyl, P. T. & Huber, W. HTSeq–a Python framework to work with high-throughput
sequencing data. _Bioinformatics_ 31, 166–9 (2015). Article CAS Google Scholar * Kanehisa, M. _et al_. KEGG for linking genomes to life and the environment. _Nucleic Acids Res_ 36, D480–4
(2008). Article CAS Google Scholar * Mao, X., Cai, T., Olyarchuk, J. G. & Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled
vocabulary. _Bioinformatics_ 21, 3787–93 (2005). Article CAS Google Scholar * Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool.
_J Mol Biol_ 215, 403–10 (1990). Article CAS Google Scholar * Griffin, N. M. _et al_. Label-free, normalized quantification of complex mass spectrometry data for proteomic analysis. _Nat
Biotechnol_ 28, 83–9 (2010). Article CAS Google Scholar * Team, R. RStudio: Integrated Development for R. (RStudio, Inc., Boston, MA, 2015). Download references ACKNOWLEDGEMENTS We
acknowledge funding support from the Natural Science Foundation of Guangdong Province [2015A030312005], the National Natural Science Foundation of China [31672611] and the Science and
Technology Program of Guangzhou [201607010177]. We thank Applied Protein Technology (aptbiotech.com) for their assistance with the proteomic analysis. AUTHOR INFORMATION Author notes * Yu
Sun and Jikai Wen contributed equally. AUTHORS AND AFFILIATIONS * Guangdong Provincial Key Laboratory of Protein Function and Regulation in Agricultural Organisms, College of Life Sciences,
South China Agricultural University, Guangzhou, Guangdong, 510642, P.R. China Yu Sun, Jikai Wen, Ruohong Chen & Yiqun Deng * Key Laboratory of Zoonosis of Ministry of Agriculture and
Rural Affairs, South China Agricultural University, Guangzhou, Guangdong, 510642, P.R. China Yu Sun, Jikai Wen, Ruohong Chen & Yiqun Deng Authors * Yu Sun View author publications You
can also search for this author inPubMed Google Scholar * Jikai Wen View author publications You can also search for this author inPubMed Google Scholar * Ruohong Chen View author
publications You can also search for this author inPubMed Google Scholar * Yiqun Deng View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS Y.S.,
J.W. and Y.D. planned and initiated the study. J.W. and R.C. performed laboratory work. Y.S. performed the computational analyses. All authors contributed to data interpretation and
manuscript completion. CORRESPONDING AUTHOR Correspondence to Yiqun Deng. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION
PUBLISHER’S NOTE: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLIMENTFIGURESTABLES
RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and
reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes
were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If
material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain
permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS
ARTICLE Sun, Y., Wen, J., Chen, R. _et al._ Variable protein homeostasis in housekeeping and non-housekeeping pathways under mycotoxins stress. _Sci Rep_ 9, 7819 (2019).
https://doi.org/10.1038/s41598-019-44305-0 Download citation * Received: 25 January 2019 * Accepted: 13 May 2019 * Published: 24 May 2019 * DOI: https://doi.org/10.1038/s41598-019-44305-0
SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to
clipboard Provided by the Springer Nature SharedIt content-sharing initiative