Semisynthesis of 5-O-ester derivatives of renieramycin T and their cytotoxicity against non-small-cell lung cancer cell lines
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

The semisynthesis of 5-O-ester derivatives of renieramycin T was accomplished through the photoredox reaction of renieramycin M (1), a bistetrahydroisoquinolinequinone alkaloid isolated from
the Thai blue sponge Xestospongia sp. This process led to the conversion of compound 1 to renieramycin T (2), which was subsequently subjected to Steglich esterification with appropriate
acylating agents containing linear alkyl, N-tert-butoxycarbonyl-L-amino, and heterocyclic aromatic substituent. Notably, the one-pot transformation, combining the photoredox reaction and
esterification led to the formation of 7-O-ester derivatives of renieramycin S due to hydrolysis. Subsequently, the in vitro cytotoxicity of the 17 semisynthesized derivatives against human
non-small-cell lung cancer (NSCLC) cells in parallel with normal cell lines was evaluated. Among the tested compounds, 5-O-(3-propanoyl) ester of renieramycin T (3b) exhibited potent
cytotoxic activity with half-maximal inhibitory concentration (IC50) values at 33.44 and 33.88 nM against H292 and H460 cell lines, respectively. These values were within the same range as
compound 1 (IC50 = 34.43 and 35.63 nM) and displayed twofold higher cytotoxicity compared to compound 2 (IC50 = 72.85 and 83.95 nM). The steric characteristics and aromatic orientation of
the 5-O-ester substituents played significant roles in their cytotoxicity. Notably, derivative 3b induced apoptosis with minimal necrosis, in contrast to the parental compound 1. Hence, the
relationship between the structure and cytotoxicity of renieramycin–ecteinascidin hybrid alkaloids was investigated. This study emphasizes the potential of the series of 5-O-ester
derivatives of renieramycin T as promising leads for the further development of potential anti-NSCLC agents.
Lung carcinoma represents a major public health problem that has become one of the leading causes of cancer incidence and mortality worldwide1. Behavioral risks such as smoking in
conjunction with factors including age, gender, genetics, environment, and air pollution contribute as the main risk factors in the development of lung tumors2,3,4. Non-small-cell lung
cancer (NSCLC) stands as the most prevalent type, accounting for 85% of all lung cancer cases5. Treatment approaches for NSCLC encompass chemotherapy, radiation therapy, immunotherapy,
targeted therapy, and personalized therapy5, 6. However, due to poor prognosis, a low 5-year survival rate, and challenges related to drug resistance5, ongoing research efforts are devoted
to the advancement of NSCLC treatment.
Marine natural products characterized by their distinctive chemical structures and biological activities have emerged as promising sources for potential anticancer drug leads7,8,9,10. Among
these compounds, 1,2,3,4-tetrahydroisoquinoline (THIQ) alkaloids belonging to the saframycin family, including saframycins, renieramycins, safracins, ecteinascidins, and their synthetic
derivatives, have demonstrated notable therapeutic impacts in chemotherapy11, 12. Trabectedin (or Ecteinascidin 743), which contains THIQ moieties as a fused-ring core structure, was
isolated from the Caribbean tunicate Ecteinascidia turbinata, and received approval from the United States Food and Drug Administration in 2015 for the treatment of soft tissue sarcoma13.
Moreover, lurbinectedin (PM01183), a tetrahydropyrroloquinoline analog of trabectedin exhibiting enhanced antitumor activity, was approved in 2020 as a the second-line treatment of
metastatic small-cell lung cancer14, 15. Anticancer mechanism of trabectedin involves the DNA alkylation by iminium ion at C–21 to generate the permanent covalent bond at the N2 position of
guanine within the DNA minor groove16, 17. Additionally, trabectedin has been reported to induce apoptosis in human anaplastic large cell lymphoma (JB6) cells through p53 and caspase 3
pathways18.
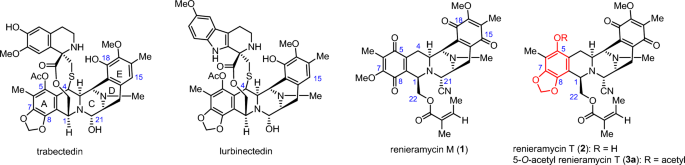
Renieramycin M (1) and renieramycin T (2) are the THIQ products isolated from the Thai blue sponge Xestospongia sp. with 1 being the major compound and 2 the minor compound19, 20. Both
compounds possess a fused pentacyclic core structure (ring A–E). Compound 1 is classified as a bistetrahydroisoquinolinequinone alkaloid, while compound 2 is categorized as a
renieramycin–ecteinascidin hybrid alkaloid, featuring a 1,3-dioxole moiety on ring A, similar to the known chemotherapeutic drugs trabectedin and lurbinectedin (Fig. 1). Compound 2 was
successfully semisynthesized from compound 1 through ambient-light-induced intramolecular cyclization in its marine natural habitat. This process was supported by studies demonstrating an
efficient intramolecular photoredox reaction, which smoothly converted 7-methoxy-6-methyl-1,2,3,4-tetrahydroisoquinoline-5,8-dione moiety on ring A of compound 1 into a
5-hydroxy-tetrahydroisoquinol-1,3-dioxole moiety of compound 2 in excellent yields21,22,23,24.
The structure of 1,2,3,4-tetrahydroisoquinoline and bistetrahydroisoquinolinequinone marine alkaloids.
Various studies have reported the structure–cytotoxicity relationship studies of compound 1 and its semisynthetic analogs against the metastatic human H292 and H460 NSCLC cell lines25, 26.
The underlying mechanisms of anticancer activity for compound 1 involve the induction of apoptosis in lung cancer cells through the p53-dependent pathway27, 28, including
mitochondria-dependent pathway29. Additionally, compound 1 has been found to suppress the levels of anti-apoptotic proteins, namely myeloid cell leukemia-1 (MCL-1), and sensitized
anoikis-resistant lung cancer cells to anoikis30. Furthermore, compound 1 exhibits potent anti-metastatic property by inhibiting epithelial-to-mesenchymal transition (EMT) in lung cancer31,
along with the suppression of lung cancer stem cell (CSC) markers32. In contrast, limited studies have been conducted on compound 2 and its semisynthetic analogs against NSCLC have not yet
been deeply conducted because of its limited quantity in natural sources20, 33. Initially, the naturally derived 2 was chemically modified by esterification to obtain only two derivatives
including 5-O-acetyl renieramycin T (3a)34 and 5-O-(N-Boc-L-alanine)-renieramycin T35 (Boc: tert-butoxycarbonyl). Pharmacological insights into the renieramycin–ecteinascidin hybrid
alkaloids have provided valuable information regarding their anti-lung cancer properties. Compound 3a induces cell death in H292 lung cancer cells through p53-dependent apoptosis, involving
the suppression of the antiapoptotic B-cell lymphoma-2 protein, and reduction of the proapoptotic Bax protein34. Moreover, 5-O-(N-Boc-L-alanine)-renieramycin T induced spheroid formation and
apoptosis in lung cancer cells, while inhibiting CSC signals by suppressing the Akt protein35.
Compounds 1, 2, and 5-O-acetyl renieramycin T (3a) have demonstrated potent cytotoxicity with nanomolar half-maximal inhibitory concentrations (IC50) against several human cancer cell lines,
including colon, lung, pancreatic, and breast cacinomas20. Compound 1 demonstrates promising anticancer effects by inducing apoptosis in lung cancer cells via the p53-dependent pathway27.
However, the presence of two quinone moieties, compound 1 induces accidental necrosis and increases the reactive oxygen species levels on the lung cancer H23 cell line. Nevertheless, a
targeted modification aimed at one quinone group to form the 5-O-acetylated hydroquinone significantly diminishes the unintended necrotic effect of the parent compound 1 on lung cancer H23
cells36. Consequently, compound 1 has undergone chemical modifications, including hydrogenation, esterification, and air oxidation, to yield diverse 5-O-ester monohydroquinone analogs of
renieramycin M, enabling the investigation of structure–cytotoxicity relationships in NSCLC25, 26, 36,37,38. Notably, compound 2, an alkaloid possessing a tetrahydroisoquinolinequinone
structure with a 1,3-dioxole motif on ring A similar to the chemotherapeutic drug trabectedin, predominantly induces apoptosis-mediated cell death39. These findings provide compelling
evidence that modifying the renieramycin core structure to feature a solitary quinone group while incorporating the 1,3-dioxole motif on the ring A system holds significant potential in
terms of its anti-lung cancer efficacy. Specifically, it promotes programmed cell death through apoptosis while reducing unintended necrosis.
Based on the reported data, a renieramycin–ecteinascidin hybrid alkaloid exhibits potent cytotoxicity, a unique anticancer mechanism devoid of unwanted toxicity, and benefits from a mild
synthetic approach. Thus, in the present study, a novel series of 5-O-ester derivatives of 2 was semisynthesized by the intramolecular photoredox reaction of 1 followed by Steglich
esterification with suitable acylating agents, including linear alkyl, N-tert-butyl-carbamate-containing amino acid, and heterocyclic aromatic substituents. Furthermore, a one-pot protocol
for the synthesis of 7-O-ester derivatives of renieramycin S was investigated. Next, the in vitro cytotoxic of the 5-O-ester derivatives of 2 was evaluated against highly metastatic human
NSCLC cell lines (H292 and H460) along with the normal cell lines including dermal papilla (DP), human keratinocyte (HaCaT) and non-tumorigenic bronchial epithelial (BEAS-2B) cell lines by
using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. In addition, compounds 1 and 3b were preliminarily investigated the mechanisms of cell death using a
Hoechst 33342 and propidium iodide (PI) nuclear co-staining assay. The effect of compounds 1 and 3b on apoptosis and necrosis was confirmed by flow cytometric analysis using Annexin V
FITC/PI double staining. Therefore, this study aims to elucidate the structure–cytotoxicity relationship of compound 2 and its semisynthetic derivatives, which can contribute to the
development of biologically active tetrahydroisoquinoline marine natural products as potential cytotoxic agents.
Compound 1 was isolated from Xestospongia sp. collected from Si-Chang Island, Thailand, using a previously reported protocol. The extraction process involved pretreatment with a 10%
potassium cyanide solution in a pH 7 phosphate buffer, followed by methanolic extraction19. The resulting compound 1 was obtained as a stable orange solid and served as a precursor for the
mild and regioselective semisynthesis of 2 and its ester derivatives. This semisynthesis involved a two-step chemical modification process consisting of intramolecular photoredox
transformation and Steglich esterification (Fig. 2). The naturally derived 1 was irradiated by an 18 W fluorescent lamp for 24 h to form a 1,3-dioxole moiety at C–7 and C–8 via light-induced
radical formation, followed by intramolecular cyclization to obtain 2 in an excellent yield21,22,23,24.
Without purification to avoid unexpected hydrolysis, compound 2 was further reacted with a suitable acylating agent in the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI), a
water-soluble carbodiimide coupling reagent, and 4-dimethylaminopyridine (DMAP), a nucleophilic base catalyst, for the Steglich esterification40 to produce a new series of 5-O-ester
derivatives having linear alkyl (3a and 3b), N-Boc-L-amino (3c–3f), and heterocyclic aromatic (3g–3o) esters. Overall, the chemical modification of 1 into 15 ester derivatives of 2 proceeded
smoothly, with acceptable to good yields (31–81%, based on 2 recovery). The reactivity of the acylating agents was controlled by their steric and electronic properties. The chemical
transformation was improved by increasing the equivalents of EDCI and DMAP. Interestingly, performing photoredox reaction and Steglich esterification simultaneously as a one-pot procedure
led to the transformation of 1 into 7-O-ester derivatives of renieramycin S41. The studies were conducted by using propionyl chloride and nicotinic acid as the acylating agent to obtain
compounds 4a and 4b, respectively as the major products. The proposed mechanism involved hydrogen abstraction, electron transfer, hydrolysis, and esterification24, 41.
The chemical structures of the semisynthetic compounds (3a–3o, 4a, and 4b) were fully elucidated by extensive spectroscopic analyses, including nuclear magnetic resonance (NMR) spectroscopy,
high-resolution mass spectroscopy, infrared (IR) spectroscopy, and electronic circular dichroism (ECD) (see Supporting Information). The characteristic proton chemical shifts of the
5-O-ester derivatives, specifically the methylenedioxy moiety at the newly formed 1,3-dioxole ring fused with ring A were observed as a pair of doublets at 6.01 ± 0.05 ppm. Moreover, the
characteristic carbon chemical shifts (δC) at the C–5 quaternary carbon, the OCH2O motif located between C–7 and C–8, and the carbonyl moiety of quinone at C–15 and C–18 on ring E appeared
at 140.1 ± 0.8, 101.9 ± 0.02, 185.5 ± 0.4, and 182.4 ± 0.4 ppm, respectively. Note that the C–5 quaternary carbon signal of the 5-O-ester derivatives was shifted upfield compared to compound
2, which contains a 5-hydroxyl group. The signals corresponding to the additional ester substituents were consistent with their respective chemical structures. The resulting ester carbonyl
moiety at C–1′ showed signals at 163.7 ± 7.6 ppm.
Furthermore, the formation of the 1,3-dioxole motif at ring A was confirmed by heteronuclear multiple bond correlations (HMBCs) between the methylenedioxy proton and the aromatic carbons at
C–7 and C–8, as well as between the methine proton at C–1 and the carbon signal from C–8 (Fig. 3). The presence of the additional ester motif at C–5 was supported by HMBCs between the proton
signal of the acyl group and the carbon signal of the methyl group at C–6, which were detected in compounds 3a and 3b. Furthermore, compounds 4a and 4b showed carbon signals corresponding
to four carbonyl moieties on bistetrahydroisoquinolinequinone (rings A and E) and the methoxy group at C–17, similar to 119 and renieramycin S42. The signals of the propanoyl and
3-pyridinecarbonyl ester substituents of 4a and 4b were observed, whereas the signals of the methylenedioxy group were absent. According to the HMBCs, the additional ester substituent at C–7
of 4a was confirmed by the carbon signal of the carbonyl group (C–1′) and the proton signal of the methyl group at C–6. Besides, the long-range correlations were observed between the
methylene proton at C–14 and the carbon at C–15, as well as between the methine proton at C–11 and the carbon at C–18, in all derivatives. These correlations indicated the presence of a
quinone skeleton on ring E.
Heteronuclear multiple bond correlations (HMBCs) in the 5-O-ester derivatives of renieramycin T and 7-O-ester derivatives of renieramycin S.
The in vitro cytotoxicity of all the semisynthesized esters and their precursors against H292 and H460 human NSCLC cell lines was analyzed based on the mitochondrial dehydrogenase activity
via the MTT assay43 (Table 1). The compounds were tested at doses of 1–250 nM. Cisplatin and doxorubicin, which are the standard chemotherapeutic drugs for the treatment of NSCLC, were used
as positive controls. The series of 5-O-ester derivatives of renieramycin T, 3a–3o, showed potent cytotoxicity in nanomolar concentrations against both cell lines. Moreover, the mother
compound 1 exhibited strong cytotoxicity (IC50 of 34.43 ± 1.70 and 35.63 ± 1.82 nM against the H292 and H460 cell lines, respectively). However, the extended use of 1 is limited by the
concerns for unwanted necrosis, which is an unprogrammed form of cell death caused by the presence of two quinone moieties36. Alkaloid 2, which is a highly oxygenated aromatic compound
containing a methylenedioxy bridge at ring A and a quinone moiety at ring E, exhibited IC50 values of 72.85 ± 2.88 and 83.95 ± 3.63 nM against the H292 and H460 cell lines, respectively.
Although it showed approximately a twofold weaker cytotoxicity than compound 1, the further investigation of 5-O-ester derivatives of renieramycin T would provide an initial
structure–cytotoxicity relationship study to investigate the promising cytotoxic agents designed to diminish unwanted toxicity. Among the derivatives having 5-O-alkyl ester and 5-O-amino
ester substituents, compounds 3a–3d possessed impressive cytotoxicity similar to 1 against the H292 (IC50 from 33.44 ± 0.87 to 43.43 ± 3.00 nM) and H460 (IC50 from 33.03 ± 1.55 to 41.63 ±
4.15 nM) cell lines. Derivatives 3b, which contain 5-O-propanoyl ester substituent, displayed the topmost potent cytotoxicity with IC50 of 33.44 ± 0.87 and 33.88 ± 1.95 nM against the H292
and H460 cell lines, twofold stronger than that of 2. According to the structure–cytotoxicity relationship data of the derivatives with amino ester substituents at C–537, 38, the smaller
steric substituents generally exhibited stronger cytotoxicity than the larger ones as the potency based on IC50 followed the order 3d > 3e > 3f. The 5-O-heterocyclic aromatic ester
derivatives (3g–3o) exhibited interesting cytotoxicity with nitrogen-containing aroyl substituents. Compound 3h, with 4-pyridinecarbonyl ester, showed potent cytotoxicity, with IC50 of 35.27
± 1.09 and 35.65 ± 1.64 nM against the H292 and H460 cell lines, respectively. Compound 3k, containing 3-pyridineacryloyl ester, exhibited pre-eminent cytotoxicity, with IC50 of 36.29 ±
2.06 and 38.75 ± 7.25 nM against the H292 and H460 cell lines, respectively. Interestingly, 5-O-(2-quinolinecarbonyl) ester (3i) showed twofold stronger cytotoxicity than
5-O-(3-quinolinecarbonyl) ester (3j), although they are regioisomers. Compounds 3e and 3j displayed cytotoxic profiles similar to 2. The five-membered heterocyclic ester derivatives 3l–3o,
containing indole, furan, thiophene
, and pyrrole moieties, exhibited decreased cytotoxicity. Among them, 5-O-(2-thiophenecarbonyl) ester (3n) had the weakest cytotoxicity, with IC50 of 136.83 ± 6.30 and 122.83 ± 4.99 nM
against the H292 and H460 cell lines, respectively. The IC50 values of the 5-O-ester derivatives 3a, 3b, 3d, 3h, 3i, and 3k were in the same range as the mother compound 1. Slight reductions
in cytotoxicity were observed with compounds 3c, 3f, 3g, 3j, 3l, 3m, and 3o. Notably, all tested compounds exhibited greater cytotoxicity compared to cisplatin. Compounds 3a, 3c, 3i, and 3m
demonstrated IC50 values equivalent to doxorubicin, while 3b, 3d, 3h, and 3k exhibited improved cytotoxicity compared to doxorubicin. These findings suggest that the steric property,
aromatic orientation including the substituted position of nitrogen in heterocyclic aromatic motifs and the ring size play important roles in improving the cytotoxicity37.
Interestingly, the renieramycin-type derivatives 4a and 4b, which contained linear and aromatic nitrogen heterocyclic ester substituents exhibited significantly decrease in cytotoxicity,
with reductions of 3- and fivefold against the H292 and H460 cell lines, respectively. These findings highlight the crucial role of the renieramycin–ecteinascidin hybrid core structure as an
essential pharmacophore, ensuring the maintenance of cytotoxic potency and essential interactions with pharmacologically related biomolecular targets16, 17.
The cytotoxicity of the 5-O-ester derivatives of renieramycin T and their parent compounds was assessed against normal cell lines, including dermal papilla (DP) cell line (Table 1), as well
as human keratinocyte (HaCaT) and the non-tumorigenic bronchial epithelial (BEAS-2B) cell lines (see Supporting Information, Table S1). The findings revealed that all renieramycin-type
compounds exhibited cytotoxicity within the nanomolar range when tested against the normal cell lines, demonstrating the stronger cytotoxic potency in comparison to H292 and H460 cell lines.
Notably, compounds 1 demonstrated robust cytotoxicity against the normal cell lines, with an IC50 of 7.07 ± 1.50 nM observed against the DP cell line. Almost all 5-O-ester derivatives of
renieramycin T possessed significantly reduced cytotoxicity on DP cell in comparison to the mother compounds 1. Among the series of renieramycin T derivatives, compounds 3c, 3h, 3i, and 3k
exhibited cytotoxicity levels equivalent to the compounds 1. Furthermore, the results indicated that treating the cytotoxic agents over an extended period (72 h) led to a stronger IC50 value
compared to shorter durations of treatment (10 and 24 h) (Table S1 and Fig. 4A,B). However, the heightened sensitivity of cytotoxicity against normal cell lines is a commonly observed
phenomenon with chemotherapeutic drugs such as cabazitaxel44, erlotinib45, and elotuzumab46. Regarding the in vitro cytotoxic assay against both cancerous and normal cell lines, the
5-O-(3-propanoyl) ester of renieramycin T (3b) exhibited significant cytotoxicity, indicating its potential utility for in-depth exploration of anti-NSCLC mechanisms.
Cell death modes of Renieramycin M (1) and 5-O-(propanoyl) ester derivative of renieramycin T (3b). (A) Percentages of cell viability of untreated (control) or treated NSCLC cells with
varying dosages of 1 and 3b (0–20 µM) for 10 and 24 h were represented and investigated by MTT assay. (B) IC50 values of 1 and 3b against H292, H460 cell lines were calculated compared to
the untreated control. (C–F) Morphologies of apoptotic and accidental necrotic cells on H292 and H460 stained with Hoechst 33342 and propidium iodide (PI) were captured using a fluorescence
microscope. The percentage of cell death were calculated based on the stained image in H292 and H460 cells. Data were presented as the means of triplicate samples ± SD (n = 3). The cell
death of the compound-treated cells was compared to that of the untreated controls; * = p