Intrauterine hyperglycemia increases insulin binding sites but not glucose transporter expression in discrete brain areas in term rat fetuses
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Diabetic pregnancy results in several metabolic and hormonal disorders, both in the embryo and the fetus of different species, including humans. Insulin is a potent modulator of
brain development and is suggested to promote the differentiation and maturation of hypothalamic or related extrahypothalamic structures, which are directly involved in neural inputs to the
pancreas. Because these structures are known to be specifically responsive both to insulin and glucose, we examined the effects of 48-h hyperglycemic clamps in unrestrained pregnant rats on
insulin binding and glucose transporter expression in hypothalamic and extrahypothalamic-related areas of their fetal offspring. The main result was an increase in insulin binding in the
ventromedial hypothalamic nucleus (VMH), the arcuate nucleus (AN), and the lateral hypothalamus (LH), and in the nucleus of the tractus solitarius (NTS) for extrahypothalamic areas (+30% in
the VMH, +37% in the AN, +25.8% in the LH, and +37.3% in the NTS). The deleterious effect of brain hyperinsulinism during the late gestational stage does not seem to act through glucose
transporter (GLUT) expression, inasmuch as no relationship between GLUT level and hyperinsulinism in brain areas could be observed. The specific increase in insulin binding in areas involved
in the nervous control of metabolism could be a factor in the increased glucose intolerance and impairment of insulin secretion that was previously observed in the adult rats from
hyperglycemic mothers. SIMILAR CONTENT BEING VIEWED BY OTHERS ONTOLOGY OF THE APELINERGIC SYSTEM IN MOUSE PANCREAS DURING PREGNANCY AND RELATIONSHIP WITH Β-CELL MASS Article Open access 29
July 2021 GESTATIONAL HYPERGLYCAEMIA IMPACTS GLUCOSE CONTROL AND INSULIN SENSITIVITY IN MOUSE OFFSPRING Article Open access 28 February 2025 BRAIN INSULIN SIGNALLING IN METABOLIC HOMEOSTASIS
AND DISEASE Article 09 June 2021 MAIN Diabetic pregnancy can lead to glucose intolerance and pancreatic dysfunction in offspring when they reach adult age (1–3). This situation seems to be
mainly related to intrauterine hyperglycemia, inasmuch as mild hyperglycemia induced by glucose infusion during late gestation in normal rats is sufficient to induce persistent impairment of
glucose regulation and insulin secretion in the adult offspring born of hyperglycemic mothers (4, 5). Interestingly, in this model, insulin deficiency was due to a perturbation of the
control of insulin secretion by the ANS and not to an intrinsic pancreatic β-cell defect (5). The activity of the ANS is under the influence of hypothalamic nuclei (PVN, VMH, and LH),
themselves connected to preganglionic nuclei in the brainstem (6, 7). These areas present a neuronal population able to respond electrophysiologically to insulin and glucose (8, 9). Insulin,
insulin receptors, and glucose transporters are present in the brain (8, 10). The defect in insulin secretion observed in the offspring of hyperglycemic mothers could be linked to impaired
glucose and/or insulin sensing in the brain. We hypothesized that fetal hyperglycemia and/or hyperinsulinemia could hamper the functional development of glucose and/or insulin-sensitive
brain areas. In fact, previous studies in models of perinatal hyperinsulinism (where hypothalamic insulin binding was increased) have demonstrated that hypothalamic nuclei were structurally
altered in adult life (11, 12). We first addressed the question of whether hyperglycemia and/or hyperinsulinemia during late pregnancy alter insulin binding in fetal brain areas involved in
the autonomic control of insulin secretion and glucose homeostasis. Insulin controls glucose transport in insulin-sensitive tissues through GLUT4 (10). Some studies have reported that, in
the brain, insulin stimulates glucose transport _in vitro_ in glial cells (13). _In vivo_, hyperglycemia and hyperinsulinemia lead to increased glucose utilization in the hypothalamus (14).
Altogether, these data suggest that insulin might be involved in glucose transport in some specific brain areas. Among the neural GLUT identified, GLUT1 (endothelial, glial cells) and GLUT3
(neuronal cells) are the main isoforms (10). We have recently demonstrated that GLUT2 and GLUT4 were also, at lower levels, expressed in adult hypothalamic and spinal cord areas,
respectively, in astrocytic and neuronal cells (15, 16). Modifying the GLUT2 expression in the brain led to an abnormal control of energy metabolism and deranged insulin secretion (17).
Moreover, in physiopathological model such as Zücker rats, GLUT4 expression was altered in hypothalamic nuclei (18). In the second part of the study, we focused on the fact that a possible
consequence of an alteration in insulin binding to specific brain areas could result in the modifications of expression of isoforms GLUT1, GLUT2, GLUT3, and GLUT4 in these areas during late
development in the rat fetus. The study was performed on rat fetuses at term from mothers made mildly hyperglycemic during the last 2 d of pregnancy by continuous glucose infusion. METHODS
ANIMALS. All rats were treated in accordance with European Community guidelines, and the experimentation was approved by our local institution. Three-month-old pregnant female Wistar rats
weighing 250 g were used. They were allowed free access to water and standard laboratory chow pellets (UAR 113, Usine d'Alimentation Rationnelle, Villemoisson, France). At d 19 of
pregnancy, a catheter was implanted under intraperitoneal pentobarbital anesthesia (125 mg/kg) in the right atrium _via_ the jugular vein. The technique (19) for long-term infusion in
unrestrained rats was used for glucose infusion (hypertonic 30% sterile glucose, Chaix & Du Marais, Paris, France). The infusion period started on d 2 after surgery and lasted 2 d. The
initial infusion rate was 30 μL/min. Control rats were infused with 0.9% NaCl. Blood glucose of infused rats was measured twice daily in samples drawn from caudal vessels. Glycemia was
immediately measured using a glucose analyzer (LifeScan, Milpitas, CA, U.S.A.). These controls allowed us to adjust glucose flow rate throughout the infusion to maintain mild hyperglycemia.
After centrifugation, the remaining plasma was stored at −20°C until insulin assay. Fetal blood samples originating from axillary vessels were used for measurements of both glycemia and
insulinemia. Plasma immunoreactive insulin was measured by a RIA kit (DiaSorin, Saluggia, Italy). At 21.5 d of pregnancy (normal term, 22 d), the infusions were disrupted and the fetuses
were immediately removed after cesarean section. They were successively exteriorized from the uterus, leaving placenta and umbilical cord _in situ_; blood samples were taken from the
axillary vessels, and the brains were rapidly removed and placed into freezing isopentane (−40°C) for 40 s. Fetal brains were stored until cryosection (20). BRAIN REGION DISSECTION. Fetal
brains were removed and frozen by dry ice. Coronal sections (300 μm) were obtained using a cryostat and placed upon chilled microscope slides. Whole hypothalamus and rostral spinal cord were
microdissected and used for either Northern blot, qcRT-PCR, or Western blot. QUANTIFICATION OF BRAIN INSULIN BINDING SITES. The regional binding sites were localized and quantified by means
of autoradiography (Biocom densitometer, Biocom, les Ulis, France) as already described (20). Autoradiograms were obtained by apposition of the sections on tritiated ultrafilm
(LKB-ultrafilm, Amersham Biosciences AB, Uppsala, Sweden) over 7 d. Six to 10 readings were made for each discrete structure in each animal. NORTHERN BLOT. Frozen hypothalamic and
extrahypothalamic tissues were used to extract total RNA using guanidine thiocyanate (21). Brain mRNA was further purified using an mRNA extraction kit (Amersham Biosciences AB) and the
concentrations measured by absorbance at 260 nm. A Northern blot was performed, as described (22), using 20 μg of purified mRNA. The blots were hybridized with 32P-labeled probe using a
Megaprime labeling system kit (Amersham Biosciences AB). The probes for GLUT1, GLUT3, and GLUT4 corresponded to the 541–1623, 597–1411, and 334–979 cDNA sequences, respectively, and were
constructed using PCR. Blots were exposed at −80°C with intensifying screens. Autoradiograms were scanned with an SI densitometer (Molecular Dynamics, Sunnyvale, CA, U.S.A.) and signals were
quantified using ImageQuant software (Molecular Dynamics). WESTERN BLOTS. Total membranes were isolated from homogenized brain regions (_i.e._ hypothalamus and rostral spinal cord). A total
membrane pellet was obtained by centrifugation at 10,000 _g_ for 10 min at 4°C, then the supernatant was ultracentrifuged at 180,000 _g_ for 35 min at 4°C. The membrane pellet was
resuspended in a PBS buffer containing protease inhibitors, and protein content was determined (Bio-Rad S.A., Marnes-la-Coquette, France). Samples were stored at −80°C until assays.
Fifty-microgram membrane samples were analyzed for GLUT content by classical Western blot analysis with the rabbit polyclonal GLUT1 (1/15,000) antibody (purchased from Biogenesis, Argene,
France) and the goat polyclonal GLUT3 and GLUT4 antibodies (Santa Cruz Biotechnology, Tebu, France), 1/5000 and 1/4500, respectively. The immunoblot signals were detected using the Amersham
ECL-detection kit system. Then, the nitrocellulose membranes were stripped of bound antibodies and incubated with a rabbit polyclonal antibody to β5 unit of integrin protein (Chemicon,
Temecula, CA, U.S.A.) (1/5000). Films of GLUT1, GLUT3, GLUT4, and β5-integrin were quantified as described for Northern blots. Results were normalized by quantification of corresponding
levels of β5-integrin protein. STATISTICAL ANALYSIS. Values are presented as means ± SEM. All statistical analyses were done by unpaired _t_ test with Prism software (GraphPad Software, San
Diego, CA, U.S.A.). Statistical significance was accepted at _p_ < 0.05. RESULTS PLASMA GLUCOSE AND INSULIN LEVELS IN MOTHERS AND FETUSES. As shown in Table 1, glucose infusion resulted
in sustained hyperglycemia both in glucose-infused mothers and in fetuses when compared with their respective controls. For plasma insulin (Table 1), a 5-fold increase was observed in
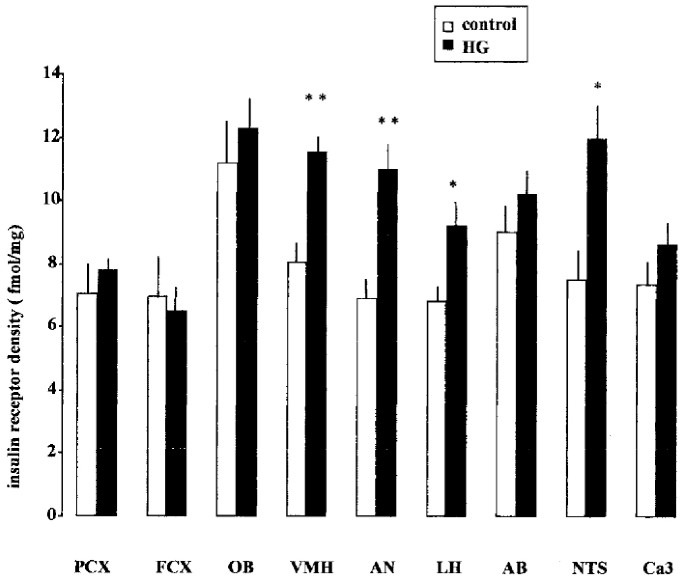
glucose-infused mothers and a nearly 7-fold increase was observed in their fetuses, compared with controls. INSULIN BINDING. Insulin receptor density was increased in several brain areas of
fetuses from hyperglycemic mothers. The greatest differences with controls were observed in the VMH, the AN, and the LH for hypothalamic areas, and in the NTS for extrahypothalamic areas
(Fig. 1). MRNA LEVELS OF GLUCOSE TRANSPORTERS. GLUT2 mRNA were undetectable in all areas studied, whatever the glycemic and insulinemic status and the methodology used, _i.e._ classic
Northern blot using mRNA preparation or the highly sensitive qcRT-PCR method (data not shown). As shown in Figure 2, exposure of fetuses to hyperglycemia and hyperinsulinemia resulted in a
marked increase in GLUT1 and GLUT3 mRNA concentrations, and a less pronounced increase in GLUT4 mRNA at the level of the rostral spinal cord (which includes the NTS). In contrast,
hypothalamic GLUT3 and GLUT4 mRNA expression was not altered in hyperglycemic fetuses. Only a slight increase of GLUT1 transcripts could be observed (Fig. 2). PROTEIN QUANTIFICATION OF
GLUCOSE TRANSPORTERS. Total membranes prepared from hypothalamic and rostral spinal cord areas of three offspring for each status (HG-HI _versus_ control) were analyzed by Western blot (Fig.
3_A_). The quantification of GLUT1, GLUT3, and GLUT4 levels in these samples, expressed as arbitrary standard units relative to the control offspring, is presented in Figure 3_B_.
Hypothalamic as well as rostral spinal cord GLUT3 and GLUT4 levels were unchanged in HG-HI fetuses when compared with controls. The 55-kD GLUT1 (endothelial isoform) was also unaffected by
the treatment, whatever the area studied. The 45-kD GLUT1 (glial and neuronal isoform) showed a significant increase, but only in the rostral spinal cord; there was no alteration in the
hypothalamus. DISCUSSION Our working hypothesis was that hyperglycemia and hyper-insulinemia during intrauterine development could act independently or together to alter the neural control
of glucose homeostasis and insulin secretion. We focused on fetal glucose transporter expression and insulin binding in brain areas involved in this regulation. The main finding of this
study is that fetuses born of hyperglycemic mothers present a significant increase in insulin binding in some brain areas, _i.e._ hypothalamic and related extrahypothalamic nuclei. This
suggests that the deterioration of glucose homeostasis and the impairment in insulin secretion in adult rats born of hyperglycemic mothers (5, 23) could at least in part originate from the
development of brain areas involved in the control of glucose homeostasis in an abnormal intrauterine milieu. Therefore, high insulin and/or glycemic levels during fetal development may
induce permanent disorders of some aspects of neuroendocrine systems. Previous studies in models of perinatal hyperinsulinism have focused on hypothalamic nuclei that are found to be
structurally altered in adulthood (11, 12). A decrease in mean area of neuronal nuclei and neuronal cytoplasm in VMH (anterior, central, and dorsomedial) and in AN were observed on d 21 of
life in offspring from diabetic rats (24). VMH alterations were accompanied by an increased number of neurons expressing TH within the AN. It has been proposed that this increased TH neuron
number might activate and overstimulate the differentiation of hypothalamic catecholaminergic systems due to hyperinsulinism. Moreover, neurons of this nucleus also express neuropeptides,
such as NPY and galanin, involved in the control of food intake and metabolism, which are altered in the adult offspring of a diabetic mothers (25). Whether an increase in insulin receptors
number could be linked to an abnormal development of mono-aminergic neurons remains to be established. Although we describe here a significantly increased density of insulin receptors in the
LH, these authors did not observe a similar disorganization in LH of hyperinsulinemic offspring (25). This could be explained by the more severe gestational diabetes status used in our
study. In addition to NTS, we found that the insulin binding sites were significantly increased. Moreover, this extrahypothalamic structure has been demonstrated to be directly linked to
hypothalamic pathways in regulating glucose levels (26, 27). In a previous study, Devaskar _et al._ (28) did not find any modification in whole brain (cerebral) insulin binding in HG-HI
fetuses, whereas the number of liver insulin receptors was largely increased. We note here the changes in insulin binding sites were restricted to hypothalamic and related extrahypothalamic
nuclei involved in the control of energety metabolism. Therefore, the results of both studies reinforce the idea that the brain targets of fetal hyperglycemia and/or hyperinsulinemia are
specifically located in nuclei including neuronal populations participating in the control of glucose homeostasis. The modifications in insulin binding in these specific areas may result in
a more general alteration in neuronal function leading to further glucose intolerance in impaired insulin secretion. Whether alterations in brain insulin binding were associated with changes
in glucose transporters of these specific structures were next investigated. Although GLUT1 and GLUT3 mRNA were increased in the hypothalamus of the HI-HG offspring, only the 45-kD GLUT1
protein, the glial form, was significantly increased in the spinal cord. Previous studies on fetal brains have focused on severe hyperglycemia, associated with hypoinsulinemia, and
quantified the protein levels of GLUT1 and GLUT3 (29). In this severe diabetic state, total fetal brain GLUT1 and GLUT3 protein were decreased, but this decrease was also present in the
streptozotocin-nondiabetic group. This suggests a chemical effect of the drug independent from maternal diabetes. The authors concluded that hyperglycemia failed to alter fetal brain glucose
transporter expression (29). Therefore, this suggests that hyperinsulinemia rather than hyperglycemia controls GLUT1 and GLUT3 expression in fetal brain. Other studies have examined the
effects of hyperglycemia on adult brain GLUT1 level without consistent results. Indeed, it has been shown that GLUT1 expression was decreased, increased, or unchanged (30). The only
modification that we observed was in the 45-kD GLUT1 glial isoform. A significant increase in this transporter was present only at the spinal cord level, where insulin binding was also
increased. Why hyperinsulinism should increase the 45-kD GLUT1 isoform only in this region without affecting the hypothalamic level is difficult to explain. To our knowledge, there are no
data describing the effect of hyperinsulinism on glucose transport in the spinal cord nuclei which can explain disturbances in nervous control of metabolism. Except for the 45-kD GLUT1 in
the spinal cord, the general absence of modification of glucose transporters in hypothalamus and spinal cord of HI-HG fetuses leads us to think that the hypothalamic alterations described
above in hyperinsulinemic fetuses might not be due to glucose transport capacity, at least at the level of the neurons and the endothelial cells that composed the blood-brain barrier. Apart
from GLUT1 and GLUT3 isoforms, we have recently demonstrated that GLUT2 and GLUT4 were expressed in adult hypothalamic and spinal cord areas (15, 16). GLUT2 mRNA are undetectable at this
age, whatever the technique used (data not shown). Regarding GLUT4, its level is not affected except for increased mRNA in the spinal cords of HG-HI offspring which did not translate to the
protein level. Such a discrepancy between mRNA and protein expression has already been reported in other tissues (31). GLUT4 mRNA and protein levels have already been described to be
regulated in developing rat brain under hypoxia or throughout embryonic development (32, 33), thus supporting the view that insulin and/or glucose are probably not the main regulators of
GLUT4 expression in the brain. The absence of modification of the insulin-dependant glucose transporter GLUT4 is concomitant with a large change in insulin binding, which is in line with the
idea of an insulin-independent GLUT4 regulation in fetal brain. Recent studies have provided evidence that the cerebral insulin pathway could act independent of glucose transport to modify
glucose homeostasis (33, 34). In summary, our study provides evidence that insulin binding is increased in hypothalamic and extrahypothalamic-related nuclei in the offspring of hyperglycemic
mothers. This increase was specific to areas involved in the nervous control of metabolism and could be a factor in glucose intolerance and impairment of insulin secretion exhibited by
young-adult rats from HI-HG mothers. The deleterious effect of brain hyperinsulinism during the late gestation does not seem to act through glucose transport expression, inasmuch as no
relationship between GLUT level and hyperinsulinism in brain areas has been observed. This does not exclude alterations in other targets of insulin action. ABBREVIATIONS * GLUT: glucose
transporter * VMH: ventromedial hypothalamic nucleus * AN: arcuate nucleus * LH: lateral hypothalamus * NTS: nucleus of the tractus solitarius * ANS: autonomic nervous system * PVN:
paraventricular nucleus * qcRT-PCR: quantitative competitive reverse transcription polymerase chain reaction * TH: tyrosine hydroxylase * HI-HG: hyperinsulinemic-hyperglycemic REFERENCES *
Freinkel N 1980 Banting lecture 1980. Of pregnancy and progeny. _Diabetes_ 29: 1023–1035. Article CAS Google Scholar * Lucas M 2001 Diabetes complicating pregnancy. _Obstet Gynecol Clin
North Am_ 28: 513–536. Article CAS Google Scholar * Van Assche FA, Holemans K, Aerts L 2001 Long-term consequences for offspring of diabetes during pregnancy. _Br Med Bull_ 60: 173–182.
Article CAS Google Scholar * Bihoreau MT, Ktorza A, Kinebanyan MF, Picon L 1986 Impaired glucose homeostasis in adult rats from hyperglycemic mothers. _Diabetes_ 35: 979–984. Article CAS
Google Scholar * Gauguier D, Bihoreau MT, Picon L, Ktorza A 1991 Insulin secretion in adult rats after intrauterine exposure to mild hyperglycemia during late gestation. _Diabetes_ 40:
109–114. Article CAS Google Scholar * Luiten PGM, Horst GJT, Koopmans SJ, Rietberg M, Steffens AB 1984 Preganglionic innervation of the pancreas islet cells in the rat. _J Auton Nerv
Syst_ 10: 27–42. Article CAS Google Scholar * Luiten PGM, Horst GJT, Steffens AB 1987 The hypothalamus, intrinsic connections and outflow pathways to the endocrine system in relation to
the control of feeding and metabolism. _Prog Neurobiol_ 28: 1–54. Article CAS Google Scholar * Baskin DG, Figlewicz DP, Woods SC, Porte D, Dorsa DM 1987 Insulin in the brain. _Ann Rev
Physiol_ 49: 335–347. Article CAS Google Scholar * Schwartz MW, Figlewicz DP, Baskin DG, Woods SC, Porte DJ 1992 Insulin in the brain: a hormonal regulator of energy balance. _Endocr Rev_
13: 387–414. CAS PubMed Google Scholar * Maher F, Vannucci SJ, Simpson IA 1994 Glucose transporter proteins in brain. _FASEB J_ 8: 1003–1011. Article CAS Google Scholar * Dorner G,
Plagemann A 1994 Perinatal hyperinsulinism as possible predisposing factor for diabetes mellitus, obesity and enhanced cardiovascular risk in later life. _Horm Metab Res_ 26: 213–221.
Article CAS Google Scholar * Plagemann A, Harder T, Rake A, Melchior K, Rittel F, Rohde W, Dorner G 1998 Hypothalamic insulin and neuropeptide Y in the offspring of gestational diabetic
mother rats. _Neuroreport_ 9: 4069–4073. Article CAS Google Scholar * Dringen R, Hamprecht B 1992 Glucose, insulin and insulin-like growth factor I regulate the glycogen content of
astroglia-rich primary cultures. _J Neurochem_ 58: 511–517. Article CAS Google Scholar * Orzi F, Lucignani G, Dow-Edwards D, Namba H, Nehlig A, Patlak CS, Pettigrew K, Schuier F, Sokoloff
L 1988 Local cerebral glucose utilization in controlled graded levels of hyperglycemia in the conscious rat. _J Cereb Blood Flow Metabol_ 8: 346–356. Article CAS Google Scholar * Leloup
C, Arluison M, Lepetit N, Cartier N, Marfaing-Jallat P, Ferre P, Penicaud L 1994 Glucose transporter 2 (GLUT 2): expression in specific brain nuclei. _Brain Res_ 638: 221–226. Article CAS
Google Scholar * Leloup C, Arluison M, Kassis N, Lepetit N, Cartier N, Ferré P, Pénicaud L 1996 Discrete brain areas express the insulin-responsive glucose transporter GLUT4. _Brain Res_
38: 45–53. CAS Google Scholar * Leloup C, Orosco M, Serradas P, Nicolaïdis S, Pénicaud L 1998 Specific inhibition of GLUT2 in arcuate nucleus by antisense oligonucleotides suppresses
nervous control of insulin secretion. _Mol Brain Res_ 57: 275–280. Article CAS Google Scholar * Alquier T, Leloup C, Arnaud E, Magnan C, Pénicaud L 2001 Altered Glut4 mRNA levels in
specific brain areas of hyperglycemic-hyperinsulinemic rats. _Neurosci Lett_ 308: 75–78. Article CAS Google Scholar * Ktorza A, Girard J, Kinebanyan MF, Picon L 1981 Hyperglycemia induced
by glucose infusion in the unrestrained pregnant rat during the last three days of gestation: metabolic and hormonal changes in the mother and fetuses. _Diabetologia_ 21: 569–574. CAS
PubMed Google Scholar * Marfaing P, Penicaud L, Broer Y, Mraovitch S, Calando Y, Picon L 1990 Effects of hyperinsulinemia on local cerebral insulin binding and glucose utilization in
normoglycemic awake rats. _Neurosci Lett_ 115: 279–285. Article CAS Google Scholar * Chomczynski P, Sacchi N 1987 Single-step method of RNA isolation by acid guanidium
thiocyanate-phenol-chloroform extraction. _Anal Biochem_ 162: 156–159. Article CAS Google Scholar * Sambrook J, Fritsch EF, Maniatis T 1989 Molecular Cloning. _A Laboratory Manual_. Cold
Spring Harbor Laboratory Press, Cold Spring Harbor, NY Google Scholar * Gauguier D, Bihoreau MT, Ktorza A, Berthault MF, Picon L 1990 Inheritance of diabetes mellitus as consequence of
gestational hyperglycemia in rats. _Diabetes_ 39: 734–739. Article CAS Google Scholar * Plagemann A, Harder T, Janert U, Rake A, Rittel F, Rohde W, Dörner G 1999 Malformations of
hypothalamic nuclei in hyperinsulinemic offspring of rats with gestational diabetes. _Dev Neurosci_ 21: 58–67. Article CAS Google Scholar * Plagemann A, Harder T, Melchior K, Rake A,
Rohde W, Dörner G 1999 Elevation of hypothalamic neuropeptide Y-neurons in adult offspring of diabetic mother rats. _Neuroreport_ 10: 3211–3216. Article CAS Google Scholar * Bray G, York
D 1979 Hypothalamic and genetic obesity in experimental animals: autonomic and endocrine hypothesis. _Physiol Rev_ 59: 719–809. Article CAS Google Scholar * Loewy AD, Haxhiu MA 1993 CNS
cell groups projecting to pancreatic parasympathetic preganglionic neurons. _Brain Res_ 620: 323–330. Article CAS Google Scholar * Devaskar SU, McMenamy K, Holtzclaw L, Sadiq F 1990
Long-term maternal-fetal exposure to high-low insulin concentrations alter liver but not brain insulin receptors. _Am J Obstet Gynecol_ 163: 1350–1356. Article CAS Google Scholar *
Schroeder RE, Rajakumar PA, Devaskar SU 1997 Effect of streptozotocin-induced maternal diabetes on fetal rat brain glucose transporters. _Pediatr Res_ 41: 346–352. Article CAS Google
Scholar * Vannucci SJ, Gibbs EM, Simpson IA 1997 Glucose utilization and glucose transporter proteins GLUT-1 and GLUT-3 in brains of diabetic (db/db) mice. _Am J Physiol_ 272: E267–E274.
CAS PubMed Google Scholar * Kahn BB 1992 Alterations in glucose transporters expression and function in diabetes: mechanisms for insulin resistance. _J Cell Biochem_ 48: 122–128. Article
CAS Google Scholar * Royer C, Lachuer J, Crouzoulon G, Roux JC, Peyronnet J, Mamet J, Pequignot JM, Dalmaz Y 2000 Effects of gestational hypoxia on mRNA levels of Glut3 and Glut4
transporters, hypoxia inducible factor-1 and thyroid hormone receptors in developing rat brain. _Brain Res_ 856: 119–128. Article CAS Google Scholar * Obici S, Zhang B, Karkanias G,
Rossetti L 2002 Hypothalamic insulin signaling is required for inhibition of glucose production. _Nat Med_ 8: 1376–1382. Article CAS Google Scholar * Alquier T, Leloup C, Atef N,
Fioramonti X, Lorsignol A, Penicaud L 2003 Cerebral insulin increases brain response to glucose. _J Neuroendocrinol_ 15: 75–79. Article CAS Google Scholar Download references
ACKNOWLEDGEMENTS The authors thank Rémy Burcelin for critical advice and Nina Crowte for editing the manuscript. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Laboratoire de Neurobiologie,
Plasticité Tissulaire et Métabolisme Energétique, UMR-CNRS 5018, IFR 31 403, CHU Rangueil, Toulouse, France Corinne Leloup, Thierry Alquier, Sanjay Mistry, Géraldine Offer, Emmanuelle Arnaud
& Luc Pénicaud * Laboratoire de Physiopathologie de la Nutrition, UMR-CNRS 7059, Paris, France Christophe Magnan, Nadim Kassis & Alain Ktorza Authors * Corinne Leloup View author
publications You can also search for this author inPubMed Google Scholar * Christophe Magnan View author publications You can also search for this author inPubMed Google Scholar * Thierry
Alquier View author publications You can also search for this author inPubMed Google Scholar * Sanjay Mistry View author publications You can also search for this author inPubMed Google
Scholar * Géraldine Offer View author publications You can also search for this author inPubMed Google Scholar * Emmanuelle Arnaud View author publications You can also search for this
author inPubMed Google Scholar * Nadim Kassis View author publications You can also search for this author inPubMed Google Scholar * Alain Ktorza View author publications You can also search
for this author inPubMed Google Scholar * Luc Pénicaud View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Corinne
Leloup. ADDITIONAL INFORMATION Supported by a grant in aid from Institut Danone. RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Leloup, C., Magnan, C.,
Alquier, T. _et al._ Intrauterine Hyperglycemia Increases Insulin Binding Sites but Not Glucose Transporter Expression in Discrete Brain Areas in Term Rat Fetuses. _Pediatr Res_ 56, 263–267
(2004). https://doi.org/10.1203/01.PDR.0000132853.35660.27 Download citation * Received: 14 August 2003 * Accepted: 12 January 2004 * Issue Date: 01 August 2004 * DOI:
https://doi.org/10.1203/01.PDR.0000132853.35660.27 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link
is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative