Dedifferentiation process driven by radiotherapy-induced hmgb1/tlr2/yap/hif-1α signaling enhances pancreatic cancer stemness
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Differentiated cancer cells reacquiring stem cell traits following radiotherapy may enrich cancer stem cells and accelerate tumor recurrence and metastasis. We are interested in the
mechanistic role of dying cells-derived HMGB1 in CD133− pancreatic cancer cells dedifferentiation following radiotherapy. We firstly confirmed that X-ray irradiation induced differentiation
of CD133− pancreatic cancer cells, from either sorted from patient samples or established cell lines, into cancer stem-like cells (iCSCs). Using an in vitro coculture model, X-ray
irradiation induced dying cells to release HMGB1, which further promoted CD133− pancreatic cancer cells regaining stem cell traits, such as higher sphere forming ability and expressed higher
level of stemness-related genes and proteins. Inhibiting the expression and activity of HMGB1 attenuated the dedifferentiation stimulating effect of irradiated, dying cells on C133−
pancreatic cancer cells in vitro and in PDX models. Mechanistically, HMGB1 binding with TLR2 receptor functions in a paracrine manner to affect CD133− pancreatic cancer cells
dedifferentiation via activating Hippo-YAP pathway and HIF-1α expression in oxygen independent manner in vitro and in vivo. We conclude that X-ray irradiation induces CD133− pancreatic
cancer cell dedifferentiation into a CSC phenotype, and inhibiting HMGB1 may be a strategy to prevent CSC enrichment and further pancreatic carcinoma relapse. SIMILAR CONTENT BEING VIEWED BY
OTHERS TARGETING LGSN RESTORES SENSITIVITY TO CHEMOTHERAPY IN GASTRIC CANCER STEM CELLS BY TRIGGERING PYROPTOSIS Article Open access 23 August 2023 THE EFFICACY OF PI3KΓ AND EGFR INHIBITORS
ON THE SUPPRESSION OF THE CHARACTERISTICS OF CANCER STEM CELLS Article Open access 10 January 2022 SELECTIVE INHIBITION OF STEMNESS THROUGH EGFR/FOXA2/SOX9 AXIS REDUCES PANCREATIC CANCER
METASTASIS Article 07 December 2020 INTRODUCTION Pancreatic carcinoma (PC) constitutes the fourth leading cause of cancer mortality1. Systemic chemotherapy or radiotherapy is regarded as an
international standard of care for advanced stage PC patients. However, more and more clinical evidence indicate that tumors develop adaptive response and become more aggressive following
radiotherapy2,3. One of the most important mechanisms for resistance is the repopulation of cancer stem cells (CSCs)4. CSCs are defined as tumor cells that express stem cell markers (cluster
of differentiation 133, CD133), have the ability to self-renew and repopulate the whole tumor. Transcription factors (including Oct4, Sox2, Nanog, and c-Myc) are known to play crucial roles
in maintaining the stemness and self-renewal abilities of cancer stem cells. CSCs play a pivotal role in tumor development, progression, and recurrence5. Current cancer treatments not only
kill a bulk amount of tumor cells but cause the expansion of CSCs population. For decades, it had been believed that the expansion of CSCs ascribed to the accelerated repopulation CSCs
following radiotherapy. Recently, more and more studies found that CSCs self-renewal and proliferation are unlikely to be the sole source of the enrichment of CSC numbers after
irradiation6,7. It has been recently reported that differentiated non-cancer stem cells may also be able to reacquire stem cell traits under ionizing radiation stress in several solid
tumors8,9. Lagadec et al. reconstituted a mixed population of breast cancer stem cells and non-cancer stem cells in vitro and confirmed that dedifferentiation of non-cancer stem cells was
the main factor for CSCs enrichment10. It is still unknown whether the dedifferentiation of non-cancer stem cells also exists in PC following chemo- or radiotherapy. Reexpression of
pluripotency factors is the main mechanisms for radiotherapy induced dedifferentiation of cancer cells in the earlier stage studies. Multiple studies demonstrated that inflammatory factors
(IL-6) can function in a paracrine or autocrine manner to affect the dedifferentiation of non-cancer stem cells11,12. Although tumor dedifferentiation is tightly regulated by genetic and
epigenetic factors, exogenous signals from the tumor microenvironments also have a very important role in this process. Dying cells and their released damage associated molecular patterns
(DAMPs) are predominant components of tumor microenvironment following radiotherapy. DAMP molecules such as calreticulin, adenosine triphosphate, or high-mobility group box 1 (HMGB1) bind to
diverse surface receptors on the neighboring cancer cells and trigger further proliferation and metastasis. However, the molecular mechanism of DAMPs in cancer cells dedifferentiation
following radiotherapy is still not clear and needs to be further investigated in detail. HMGB1 is passively released from dying or injured cells or actively secreted from cancer cells in
response to exogenous and endogenous stimuli such as hypoxia and various soluble factors (TNF-α and IL-1). HMGB1, released from tumor cells upon chemotherapy or radiotherapy, is an important
component of disordered tumor microenvironment and highly associated with cancer hallmarks including sustain proliferation, resistance to cell death, invasion and metastasis, and
recurrence13,14,15. Our previous work confirmed that dying cells derived HMGB1 accelerated pancreatic carcinoma metastasis following radiotherapy16. However, it is unclear whether HMGB1
secreted by dying cancer cells following radiotherapy is involved in expansion of CSCs, especially in the non-CSCs dedifferentiation. Therefore, we are interested if dying pancreatic cancer
cells during radiotherapy activate HMGB1-mediated paracrine signaling events that promote dedifferentiation of resident non-CSCs. To test this hypothesis, we used the Millicell coculture
system and patient-derived xenograft (PDX) tumors to show that dying pancreatic cancer cells following radiotherapy significantly induced CD133− cancer cells dedifferentiation in vitro and
tumorigenesis in vivo. Silencing HMGB1 in irradiated cancer cells (feeder cells) attenuated expression levels of stem cell-related markers (Oct4, Sox2, C-myc, and Nanog) and sphere forming
ability of CD133− reporter cells. Finally, we showed that HMGB1 functions in a paracrine manner to activate Hippo-YAP pathway and enhances hypoxia-inducible factor 1α (HIF-1α) expression in
CD133− cells in an oxygen independent manner in vitro and in vivo. MATERIALS AND METHODS CELL CULTURE AND HUMAN PANCREATIC CARCINOMA PRIMARY SPECIMENS Human pancreatic cancer cell line
(PaTu8988) was obtained from Cell Bank of China Academy of Sciences (Shanghai, China). PaTu8988 cells, a human pancreatic carcinoma of ductal cell origin, were maintained in DMEM medium
supplemented with 10% FBS, 100 U/mL penicillin, and 100 U/mL streptomycin. The cells were put in a humidified atmosphere of 95% air with 5% CO2 at 37 °C and passaged with 0.25% trypsin/EDTA
every 3 days. Cells were passaged for <2 months after recovery. Tumor specimens were obtained under a protocol approved by Department of General Surgery, the First Affiliated Hospital of
Nanjing Medical University and the Affiliated Hospital of Jiangsu University. All patients underwent fully informed consent in accordance with local research ethics committee guidelines.
Specimen site was selected to avoid both the tumor margin and necrotic core, and were kept overnight at 4 °C in DMEM with 10% FBS. Specimens were dissected into 1–2 mm3 cubes and digested in
single-cell suspension and was filtered sequentially through 100 μm cell strainers, then centrifuged at 300 × _g_ for 5 min, and washed in PBS. Human Tumor Dissociation Kit (Miltenyi
Biotec.) was used to remove contaminating stromal cells for 2 h at 37 °C. The primary cancer cells were expanded for 1 weeks and then for further use. IRRADIATION AND IN VITRO COCULTURE
SYSTEM OF CANCER CELLS Pancreatic cancer cells cultured in 6 cm dishes or Millcell insurts were irradiated at room temperature using an X-ray irradiator (Linear accelerator, Turebeam_STX,
Varian, USA) with indicated dose (2, 4, 8, 10, and 20 Gy). The dose rate of the machine is about 4 Gy/min. Corresponding controls were sham irradiated. Irradiated cells were immediately
trypsinized and reseeded for further use. Segregated irradiated cancer cells and untreated cancer cells coculture system was established as previously reported17. In brief, 5 × 104
irradiated indicated cancer cells were seeded on 0.4 μm inserts (Millicell) in DMEM with 2% FBS. After 12 h, the inserts were moved to 24-well plates containing indicated number untreated
CD133− cancer cells in DMEM with 2% FBS. Different concentration of recombinant human HMGB1 (rhHMGB1, 100, 200, 250, and 300 ng/mL) was added to the same medium above mentioned in the
inserts as positive control. Empty inserts with the same medium were used as control. The experiments were repeated three times with duplicate samples per group. FLOW CYTOMETRY AND
FLUORESCENT-ACTIVATED CELL SORTING CD133 staining was carried out as described previously18. In brief, 5 × 106 cells were harvested, disaggregated into a single-cell suspension, and
incubated with 2 mg/ml mouse anti-human CD133/phytoerythrin (PE) antibody for 30 min at 4 °C in the dark. After incubation, the samples were washed with PBS and analyzed by FACS AriaII
(Becton Dickinson, USA). For separating CD133+ and CD133− population by FACS, cultured pancreatic cancer cells growing in sphere forming media system (SFM, DMEM-F12 with 2%B27, 20 ng/ml
epidermal growth factor (EGF), 20 ng/ml basic fibroblast growth factor (bFGF), 4 ug/ml heparin, and 5 μg/ml insulin, Sigma-Aldrich) were stained for CD133. Cancer cells were incubated with
trypsin–EDTA, dissociated and passed through a 40 µm sieve. Cells were pelleted by centrifugation at 500 × _g_ for 5 min at 4 °C, resuspended in 100 µL of monoclonal mouse anti-human
CD133/PE antibody (1:10, Miltenyi Biotec.), and incubated for 30 min at 4 °C. The sorting gates were established using cells stained with isotype control PE-conjugated antibodies (BD
pharmingen). Sorted CD133+ and CD133− cells were reseeded for further use. REAGENTS TREATMENT Recombinant human (rhHMGB1, HMGBiotech, Germany) was dissolved in distilled water to make a 1000
ng/ml stock solution. When the cells grown to 80% confluency, various concentrations of rhHMGB1 (100, 200, 250, and 300 ng/mL) were added for the indicated time. The treated cells were
subjected to the following experiments. Ethyl pyruvate (EP, HMGB1 inhibitor) was purchased from MCE (USA). Cells were grown to 80% confluency, treated with EP (1:1000) for the indicated
time, and subjected to the following experiments. Stevioside (TLR2 antagonist) purchased from Topscience (Shanghai, China) and dissolved in dimethyl sulfoxide (DMSO). Cells were grown to 80%
confluency, treated with 2 μM Stevioside for the indicated time, and subjected to the following experiments. IN VITRO SPHERE-FORMING ASSAY After sorted, CD133− pancreatic cancer cells were
seeded into ultra-low adhesion plates (Corning, NY, USA) and suspended in SFM system, ranging from 1 to 256 cells/well, for 1–2 weeks to allow formation of spheres from single cells. The
culture medium was replaced by fresh medium every 2 days. After 1–2 weeks, the number and size of spheres in each well were quantified. RNAI AND GENE TRANSFECTION Pancreatic cancer cells
were seeded in six-well plates at a density of 1 × 105 cells/well to achieve a confluence of 70–80% overnight. Then, HMGB1-shRNA, TLR2-shRNA, YAP-shRNA, HIF-1α -shRNA, and negative control
shRNA (Suzhou Ribo Life Science CO., Ltd, Suzhou, China) were transfected into cells, respectively, using transfection reagent (Lipofectamine 2000, Invitrogen, China) according to the
manufacturer’s instructions. The specific shRNA sequences are listed in Supplementary Table 1. For establishing the stable sh-HMGB1 cancer cells, the lentiviral packaging kit was purchased
from Open GeneCopoeia. Lentivirus carrying HMGB1-shRNA1 and was packaged in 293T cells and concentrated from the supernatant, as instructed by the manufacturer's manual. Stable cell
lines were established by infecting lentivirus into pancreatic cancer cells followed by puromycin (1 μg/ml) selection for 10–14 days. These established stable cell lines were maintained in
DMEM containing 10% FBS and puromycin (0.75 μg/ml) for further experiments. WESTERN BLOT ANALYSIS Protein concentrations were determined by BCA method. Western blot assay was carried out as
described previously19. In brief, after extraction, proteins in the cell lysate were resolved on 4–12% Criterion XTBis-Tris gels (Bio-Rad) and transferred to a nitrocellulose membrane. After
blocking with 5% milk, membranes were incubated at 4 °C with various primary antibodies overnight. After incubation with peroxidase-conjugated secondary antibodies for 1 h at room
temperature, the signals were visualized using enhanced or super chemiluminescence and exposure to X-ray films. Antibodies against HMGB1 and β-tubulin were purchased from Abcam Company
(Cambridge, USA). Antibodies against CD133, Oct4, Sox2, Nanog, TLR2, TLR4, N-cadherin, Vimentin, E-cadherin, C-myc, p-MOB1, MOB1, YAP, p-YAP, HIF-1α, and Histone3 were obtained from Cell
Signaling Technology, Inc. (Boston, USA). The secondary antibody preparations either anti-rabbit or anti-mouse were purchased from Boster Biotechnology Company (Wuhan, China). Nuclear and
cytoplasmic proteins were isolated with Nuclear and Cytoplasmic Extraction Kit (Thermo Scientific, Waltham, MA, USA). The protein from total cells was extracted from the cultured cells,
which were homogenized in RIPA lysis buffer (1% NP40, 0.1% Sodium dodecyl sulfate (SDS), 100 μg/ml phenylmethylsulfonyl fluoride, 0.5% sodium deoxycholate, in PBS) on ice. The supernatants
were collected after centrifugation at 12000 × _g_ at 4 °C for 20 min. The separated proteins were then transferred to a PVDF membrane. The membrane blots were first probed with a primary
antibody. β-tubulin was used as a loading control for cytosol protein or total protein, and Histone3 was used as a loading control for nuclear protein. LUCIFERASE ASSAY CD133− PaTu8988
cancer cells were transiently transfected with the YAP-Luciferase/Renilla (50:1) or HRE/Renilla(50:1) reporter plasmid using transfection reagent (Lipofectamine 2000, Invitrogen,
Mississauga, Ontario, Canada) according to the manufacturer’s instructions. Luciferase assay was performed by using the Dual Luciferase Assay System kit after transfection. Activity was
assayed in three independent experiments. QUANTITATIVE REAL-TIME PCR Total RNA was extracted from cancer cells using the RNeasy kit (QIAGEN). For mRNA analysis, cDNA was synthesized from 1
μg total RNA using the Superscript III kit (Invitrogen). SYBR Green-based real-time PCR was subsequently performed in triplicate using SYBRGreen master mix (SA Biosciences) on an Applied
Biosystems Step One Plus real-time PCR machine (Thermo Fisher Scientific). For analysis, the threshold cycle (Ct) values for each gene were normalized to expression levels of GAPDH. The
primers used are listed in Supplementary Table 2. CHROMATIN IMMUNOPRECIPITATION (CHIP) ASSAY ChIP was performed according to the protocol of the chromatin immunoprecipitation assay kit.
Briefly, cells (CD133− PaTu8988 or HIF-1α knockdown CD133− PaTu8988 cancer cells) were pretreated with or without rhHMGB1 under normoxia or hypoxia condition and then cross-linked in 3.7%
formaldehyde for 15 min, quenched with glycine for 5 min, and lysed with SDS lysis buffer. Chromatin was sheared by sonication, and lysates were precleared with salmon sperm DNA/protein A
agarose slurry for 1 h and incubated with IgG or antibodies against HIF-1α or YAP in the presence of protein A agarose beads overnight. After sequential washes of the agarose beads and
eluted, the elutes were heated at 65 °C for 4 h to reverse the cross-linking and treated with RNase A for 30 min at 37 °C, followed by treatment with proteinase K for 1 h at 45 °C to remove
RNA and protein. DNA was recovered, eluted, and then assayed using PCR. The primer for the human Oct4, SOX2, C-myc, and Nanog promoter containing the HRE or TEAD site listed in Supplementary
Tables 3 and 4, respectively. CO-IMMUNOPRECIPITATION ASSAY Cells were collected and lysed in 0.5 ml lysis buffer plus protease inhibitors for 50 min on a rotor at 4 °C. After 12,000 × _g_
centrifugation for 15 min, the lysates were immunoprecipitated with 2 μg specific antibody overnight at 4 °C, and 30 μl A/G agarose beads (Santa Cruz, SC-2003) were washed and then added for
an additional 6 h. Thereafter, the precipitants were washed three times with lysis buffer, and the immune complexes were boiled with loading buffer for 5 min and analyzed by SDS–PAGE. The
following antibodies were used for immunoprecipitation: antibodies against normal Rabbit IgG [2729, CST], anti-TLR2 [D7G9Z, CST], and Anti-YAP [14074, CST]. XENOGRAFT TUMOR MODELS Animal
studies were approved by the Committee on the Use of Live Animals for Teaching and Research of the Jiangsu University. Female BALB/c nude mice (purchased from The Compare Medicine Center,
Yangzhou University, China), ages 4 weeks, were maintained under standard conditions according to institutional guidelines. Sorted CD133− PaTu8988 cancer cells were cocultured with
irradiated parental cancer cells HMGB1+ cells(HMGB1 positive, HP), irradiated HMGB1 knock down pancreatic cancer cells (iHMGB1shRNA1), rhHMGB1(150 ng/mL) and the same empty medium (PBS).
Fourteen days later, 1 × 103, 1 × 104 and 1 × 105 cells/mouse were injected into the subcutaneous of nude mice, respectively. Mice were monitored every day until the end point of day 50,
when the tumors that were palpable were taken as a positive. 1 × 106 CD133− PaTu8988 cancer cells, TLR2−CD133− PaTu8988 (TLR2 knockdown CD133− PaTu8988), YAP−CD133− PaTu8988 (YAP knockdown
CD133− PaTu8988), or HIF-1α− CD133− PaTu8988 (HIF-1α knockdown CD133− PaTu8988) cancer cells were implanted subcutaneously into the right dorsal flanks of nude mice, respectively. When the
tumors reached a volume of 200 mm3, the mice were treated with HMGB1 by peritumoral injection every 2 days for 2 weeks. Tumors were harvested 3 days after the last injection for analysis of
CD133+ cells, and expression of stem cell-related markers (Nanog and Oct4). 1 × 106 PaTu8988 cells were implanted into the nude mice. When the tumor reached a volume of 200 mm3, the mice
were received 20 Gy X-ray irradiation. Two days later, the mice were randomized to receive surrounding tumor injections of PBS, EP (HMGB1 inhibitor), Stevioside (TLR2 inhibitor),
Verteporfin(YAP inhibitor), or LW6 (HIF-1α inhibitor) for 5 days (designed for day1). The experiment was terminated on 30 day. The tumor volume and regrowth speed were monitored. PDXS AND IN
VIVO EXPERIMENTS NSG (NOD. Cg-_Prkdc__scid_ _Il2__rgtm1Wjl_/SzJ) mice were purchased from the BEIJING IDMO Co., Ltd. and maintained in Animal Center of Jiangsu University in compliance with
the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85–23, revised 1996). The experimental protocols were approved by the Committee for Ethical Affairs of Jiangsu
University (Zhenjiang, China), and the methods were carried out in accordance with the approved guidelines. All in vivo work was performed with a minimum of _n_ = 3 mice per condition.
Serial passaging of the PDX was carried out by implanting small fragments of the tumor subcutaneously into dorsal flanks of NSG mice. Experiments were performed using PDX tumors passages 4
and 5. HPCx1 and HPCx2 derived PDXs mice were fixed into special equipment and received 4Gy radiation for each 2 days and for five times. Seven days later, the mice were randomized to
receive peritumoral injections of PBS, rhHMGB1 (150 ng/ml), and HMGB1 antibody (250 ng/ml) every 2 days for 2 weeks. The experiment was terminated on 30 day. Animal weight and tumor size
were measured bi-dimensionally using calipers twice a week. IMMUNOFLUORESCENCE ANALYSES Cells were seeded on coverslips. Cells were fixed with 4% paraformaldehyde for 10 min at room
temperature, and then permeabilized with 0.1% Triton X-100. After blocking in goat serum for 1 h, slides were incubated with primary antibody (anti-YAP (1:100) and anti-HIF-1α (1:800)) for 1
h at 4 °C overnight. Slides washed three times with PBST and incubated with CY3-conjugated secondary antibodies (Invitrogen, 1:1000) for 1 h at room temperature. Slides washed three times
with PBST and incubated with DAPI for 5 min at room temperature. The slides were then washed three times with PBST and mounted. Cell images were captured with a confocal microscope (Leica).
PATIENT SELECTION The Cancer Genome Atlas (TCGA) database (https://tcga-data.nci.nih.gov/tcga; TCGA_PAAD_exp_HiSeqV2_percentile-2015-02-24TCGA pancreatic adenocarcinoma (PAAD) gene
expression by RNAseq (IlluminaHiSeq percentile) including 183 pancreatic carcinoma patient specimens was utilized to further analyse the relationship between CD133, Oct4, Nanog, Sox2, YAP,
and HIF-1α. High and low groups were defined as above and below the mean, respectively. STATISTICAL ANALYSIS All data are presented as the mean ± SEM (standard error of the mean). Linear
regression and _F_ test were used to determine the significance of TCGA data. The significances of differences between groups were analyzed using Student’s _t-_tests, one-way, or two-way
ANOVA. Values of _P_ < 0.05 were considered to be significant. All the experiments were repeated at least three times. RESULTS X-RAY IRRADIATION INDUCED CELL DEATH PROMOTES CD133−
DEDIFFERENTIATION To test the hypothesis that irradiation-induced cell death promoted cancer cell dedifferentiation, we first confirmed that CD133+ cancer stem-like cells can be enriched by
irradiation of primary pancreatic cancer cells from pancreatic carcinoma patients (P1, P2, and P3), or established pancreatic cancer cell line (PaTu8988) with clinically relevant doses in
vitro (Fig. S1A). To test if these CSCs originated from dedifferentiation of CD133− cancer cells, we cocultured lethally irradiated pancreatic cancer cells (20 Gy, lethal dose optimized in
Fig. S1B) or nonirradiated control cells (0 Gy) together with sorted CD133– cancer cells using a Millicell insert system. FACS analysis showed that compared with nonirradiated control cells,
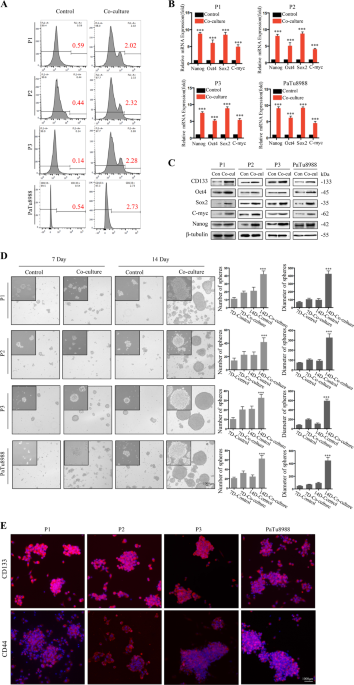
irradiated feeder cells led to more CD133+ cells dedifferentiated from CD133− cancer cells following 7 days coculture (P1: 3.4-fold induction (0.59% vs. 2.02%); P2: 5.2-fold induction
(0.44% vs. 2.32%); P3: 16.2-fold induction (0.14% vs. 2.28%); PaTu8988: 5.1-fold induction (0.54% vs. 2.73%), Fig. 1a). Furthermore, mRNA and protein expression analysis demonstrated that
stem-related markers (Nanog, Oct4, Sox2, and C-myc) were markedly increased in CD133− cancer cells following 7 days coculture with irradiated feeder cells (Fig. 1b, c). Consistently,
irradiated feeder cells induced CD133− cancer cells to form more spheres, which also presented higher levels of CSC markers CD133 and CD44 (Fig. 1d, e). Together, these results suggest that
irradiation-induced cell death promotes CD133− dedifferentiation into CSCs. IRRADIATION INDUCED CELL DEATH PROMOTES CD133− CANCER CELL DEDIFFERENTIATION VIA HMGB1-TLR2 INTERACTION We
previously found that dying cells release HMGB1 to drive an aggressive phenotype in pancreatic cancer16. To examine the possible role of dying cell derived HMGB1 in dedifferentiation of
resident cancer cells, we silenced HMGB1 using two RNAi in primary and established pancreatic cancer cells, and the silencing efficacy was confirmed by western blot (Fig. S2A). The CD133−
cancer cells were then con-cultured with following agents for 7 days: (i) Different concentration of recombinant human HMGB1(rhHMGB1, 100, 200, 250, and 300 ng/mL); (ii) 20 Gy X-ray
irradiated HMGB1 wide type cancer cells (iHMGB1+); (iii) 20 Gy X-ray irradiated HMGB1 knock down cancer cells (iHMGB1shRNA1); (iv) 20 Gy X-ray irradiated HMGB1 knock down cancer cells
(iHMGB1shRNA2); (v) 250 ng/mL rhHMGB1+ iHMGB1shRNA1; (vi) 250 ng/mL rhHMGB1+ethyl pyruvate (EP, HMGB1 inhibitor); and (vii) PBS(control). iHMGB1+ resulted in significant upregulation of mRNA
and protein expression of stem cell markers (Oct4, Sox2, and Nanog) in CD133− cancer cells, their sphere-forming ability, as well as CD133+ cell percentage 7 days post coculture, which were
comparable to 250–300 ng/mL rhHMGB1 (Fig. 2a–d). In contrast, inhibiting HMGB1 expression (iHMGB1shRNA1 and iHMGB1shRNA2) in feeder cells or activity (EP) attenuated these effects (Fig.
2a–d and Fig. S3A), supporting our hypothesis that HMGB1 released from irradiated cancer cells positively mediates this dedifferentiation process. It is well known that EMT process can
induce cancer cell dedifferentiation into CSCs. We also further confirmed that CD133− cancer cells exposure to the HMGB1+ irradiated cells or rhHMGB1 led to acquisition of higher migration
ability (Fig. S2B) and mesenchymal phenotype accompanied by up-regulation of mesenchymal makers (N-cadherin and Vimentin) and down-regulation of epithelial marker (E-cadherin) (Fig. 2b and
Fig. S3A). Assessing putative cell surface receptors (TLR2 and TLR4) binding with extracellular HMGB1, we found that TLR2, but not TLR4, was upregulated in the CD133− cancer cells when
treated with HMGB1+ irradiation cells or rhHMGB1 (Fig. 3a). TLR2 and stem cell-related markers (CD133, Oct4, Sox2 C-myc, and Nanog) were evaluated in a time and concentration dependent
manner with rhHMGB1 (Fig. 3a). Co-IP assay further confirmed the interaction between HMGB1 and TLR2 when CD133− cancer cells were cocultured with the 250 ng/ml rhHMGB1 (Fig. 3b). To study
the functional consequences of TLR2 in HMGB1 mediating CD133− cancer cells dedifferentiation, TLR2 was inhibited by shRNA technology or used the TLR2 inhibitor (Stevioside) in the CD133−
cancer cell coculture system. TLR2 inhibition exhibited pronounced suppression of stem cell markers induced by HMGB1 (Fig. 3c, d, and Fig. S3B). Consistently, HMGB1 promoted CD133− cancer
cells sphere forming ability was also abrogated following suppress the expression and function of TLR2 (Fig. 3e). In summary, these results indicate that extracellular HMGB1 interacts with
TLR2 receptor on CD133− cancer cells to promote their dedifferentiation into CSCs. HMGB1-TLR2 INDUCED CD133− CANCER CELLS DEDIFFERENTIATION VIA REGULATING HIPPO-YAP PATHWAY To identify
downstream effectors of HMGB1-TLR2 mediated cancer cell dedifferentiation, we examined the expression levels of several key signaling pathways regulating cancer cell plasticity, and noticed
the protein expression of YAP and MOB1 (core component of the Hippo pathway) was slightly enhanced upon rhHMGB1 stimulation (Fig. 4a). Interestingly, we found YAP phosphorylation(Ser397),
which is associated with YAP protein degradation, was significantly reduced in CD133− cancer cells treated with HMGB1+ irradiated cells or rhHMGB1 (Fig. 4a, c). YAP mRNA expression was not
affected regardless of HMGB1 presence, further suggesting HMGB1 enhanced YAP protein expression by inhibiting YAP phosphorylation (Fig. 4b). HMGB1decreased the YAP phosphorylation in a time
dependent manner at the peak inhibition on 6 h post treatment, in accompany with enhanced expression of YAP (Fig. 4c). Luciferase assay revealed that YAP luciferase activity was promoted
(Fig. 4d). Consistently, mRNA expression levels of YAP target genes such as cyclin E, SPP1, and CTGF were up-regulated in CD133− cancer cells following rhHMGB1 treatment (Fig. 4e). YAP
silencing reduced the expression of stem cell-related markers (CD133, Nanog, Sox2), as well as sphere-forming ability of rhHMGB1 stimulated CD133− cancer cells (Fig. 4g, h, Fig. S3C),
confirming its role in cancer cell stemness. Furthermore, TLR2 silencing enhanced the activation of YAP(the rational of ratio of phosphorylation YAP to total YAP increased) in HMGB1
stimulated CD133− cancer cells (Fig. 4f, Figs. S3B and S4), suggesting that YAP is downstream of HMGB1-TLR2 signaling to induce CD133− cancer cells dedifferentiation. YAP/HIF-1Α COMPLEX
CONTRIBUTE TO HMGB1 INDUCED CD133− CANCER CELLS DEDIFFERENTIATION In addition to acting as an oncogene in many cases, YAP may complex with other transcription factors to regulate cancer
biology including HIF-1α, which controls cancer cells stemness following radiotherapy in an oxygen dependent or independent manner. We found that protein instead mRNA level of HIF-1α rose in
CD133− cancer cells following HMGB1 treatment in normoxia (Fig. 4a–c). Knockdown of TLR2 or YAP in CD133− cancer cells decreased HIF-1α expression under normoxia condition (Fig. 4f),
suggesting HIF-1α is downstream of TLR2 and YAP. To further confirm the relationship between YAP and HIF-1α, we first examined the localization of YAP and HIF-1α in CD133− cancer cells
following rhHMGB1 treatment by immunofluorescence. YAP and HIF-1α can be detected in CD133− cancer cells with or without rhHMGB1 treatment. YAP and HIF-1α were primarily detected in the
cytoplasm of cancer cells without HMGB1 treatment, while displayed distinct nuclear localization following rhHMGB1 treatment (Fig. 5a). Western blot also confirmed that YAP and HIF-1α
nuclear translocation enhanced following rhHMGB1 treatment (Fig. 5b). Moreover, YAP nuclear translocation was significantly inhibited in HIF-1α knockdown CD133− cancer cells in the presence
of rhHMGB1 treatment (Fig. S3E). YAP-HIF-1α complex was observed in CD133− cancer cells treated with rhHMGB1 in both normoxia and hypoxia (Fig. 5c) by co-IP assay. YAP luciferase activity in
presence of HIF-1α was promoted in rhHMGB1 treated CD133− cancer cells. Moreover, HRE luciferase activity, indicated HIF-1α DNA binding ability, was enhanced in HMGB1 treated CD133− cancer
cells and suppressed in the presence of EP under hypoxia (Fig. 5d). To investigate whether HIF-1α directly regulates pluripotent genes (Oct4, Sox2, C-myc, and Nanog) expression, we searched
the promoter regions of these genes and identified site consensus to HIF-1α binding sequences (Fig. S5A). ChIP assay revealed that HMGB1 promoted HIF-1α binding to promoter regions of these
pluripotent genes, while the binding ability was abolished in HIF-1α knockdown cells in presence of rhHMGB1 (Fig. 5e). To further confirm that HIF-1α is required for the recruitment of YAP,
we assessed if HMGB1 promoted YAP binding to pluripotent genes promoter regions (Fig. S5B) by CHIP in HIF-1α knockdown and control CD133− cancer cells. The results revealed that HMGB1
promoted YAP binding to pluripotent gene promoter regions in the presence of HIF-1α, while the binding ability was abolished in HIF-1α knockdown cells in presence of rhHMGB1 (Fig. 5e, Fig.
S5B). Taken together, these data indicate that dying cells derived HMGB1 mediated formation of nuclear YAP-HIF1α complex, which further activates expression of the pluripotency factors.
HMGB1-TLR2-YAP/HIF-1Α INDUCED CD133− CANCER CELL DEDIFFERENTIATION IN VIVO In order to confirm the molecular mechanisms of HMGB1 inducing cancer cell dedifferentiation in vivo, we firstly
determined the tumor initiation potential of CD133− cancer cells in similar in vitro conditions. Sorted CD133− PaTu8988 cancer cells were cocultured with irradiated parental cancer cells
(iHMGB1+ cells), irradiated HMGB1 knockdown cancer cells (HMGB1− cells), rhHMGB1 (150 ng/mL), or normal medium (control group) and then injected subcutaneously into nude mice for the
indicated cell number (1 × 104, 1 × 105 and 1 × 106 cells/mouse were injected, respectively). CD133− cancer cells cocultured with iHMGB1+ cells and rhHMGB1 showed significantly higher tumor
initiating capacity than the other groups. 4/4 mice developed tumors in each group until the cell number reached 1 × 106, but tumors derived from HMGB1+ cells and rhHMGB1 coculture group
were larger than the other groups (Fig. 6a). To confirm the molecular mechanisms in vivo, 1 × 106 CD133− PaTu8988 cancer cells, TLR2, YAP, or HIF-1α knockdown PaTu8988CD133− cancer cells
were implanted subcutaneously into the right dorsal flanks of nude mice, respectively. When the tumors reached a volume of 200 mm3, the mice were treated with rhHMGB1 by peritumoral
injection every 2 days for 2 weeks, and tumors were harvested 3 days after the last injection for analysis of stemness. HMGB1 treatment resulted in significant upregulation of stem cell
markers (Oct4 and Nanog) and CD133+ cell proportion in fresh tumor tissues, which were abrogated by silencing TLR2, YAP, or HIF-1α (Fig. 6b, c). As a comparison to rhHMGB1 treatment, the
mice received peritumoral injections of PBS, EP (HMGB1 inhibitor), Stevioside (TLR2 inhibitor), Verteporfin (YAP inhibitor), or LW6 (HIF-1α inhibitor) every 2 day for 2 weeks. Combination
treatment of X-ray irradiation with TLR2-YAP-HIF-1α pathway inhibitors prevented tumor relapse (Fig. 6d), indicating that inhibiting the HMGB1-TLR2-YAP-HIF-1α pathway blocked
radiotherapy-induced dedifferentiation of primary tumor cells and also prevents tumor relapse. RADIATION-INDUCED CANCER CELL DEATH CONSTITUTE A SUPPORTING NICHE FOR CANCER CELL STEMNESS
Using two established patient derived xenograft (PDX) models (HPCx1 and HPCx2), we explored the role of HMGB1 in mediating the tumor relapse. The mice received 20 Gy X-ray irradiation, and
were randomized 7 days later to receive surrounding tumor injections of PBS, HMGB1, and HMGB1 antibody every 2 day for 2 weeks. The experiment was terminated on 50 days upon treatment. HMGB1
treatment promoted tumor regrowth (Fig. 7a) and increased TLR2-YAP-HIF-1α signaling activation compared with the parental control (Fig. 7b). Moreover, the HMGB1 treated model has a higher
percentage of CD133+ cancer cells, which were abrogated by HMGB1 antibody treatment (Fig. 7c). Using the information from TCGA database (https://genome-cancer.ucsc.edu, _n_ = 183), we
evaluated the correlation gene expression of CD133, Oct4, Nanog, Sox2, YAP, and HIF-1α, and the heat-map revealed a linear relationship between the expression of these markers in TCGA
protein array (Fig. 7d). DISCUSSION In this study, we confirmed CD133− pancreatic cancer cells dedifferentiation to CSCs using primary human pancreatic carcinoma tissues, established
pancreatic cancer cell line, and a PDX model following radiotherapy. Furthermore, dying cell derived HMGB1 activates TLR2/YAP/HIF-1α pathways and induces neighboring CD133− pancreatic cancer
cells dedifferentiation, which is an important signaling event underlying tumor relapse following radiotherapy (Fig. 8). Therefore, blocking this pathway may prevent both enrichment of
CD133+ cancer stem cell population and tumor recurrence. The resulting cell death may also stimulate the generation of DAMPs and their corresponding pattern recognition receptors. As one
most common DAMPs, HMGB1 is complicated in tumor treatment resistant by its paradoxical dual activities. Huang et al. reported that HMGB1 participated in immune-scavenging in advanced rectal
cancer that have undergone neoadjuvant chemo- or radiotherapy20. Yin et al. found that HMGB1-mediated autophagy attenuates gemcitabine-induced apoptosis in bladder cancer cells21.
Extracellular HMGB1 on cancer cell stemness has gathered increasing attention. Cancer-associated fibroblasts derived HMGB1 promoted stemness and tumourigenicity in breast cancer22. HMGB1 is
also found to act as a potent EMT driver in colorectal carcinoma and gastric cancer23,24. It has confirmed that cancer cells undergoing EMT acquire stem cell like properties25. These
researches indirectly confirmed the role of HMGB1 in cancer cells plasticity. Our results for the first time supply the direct evidence that dying cells derived HMGB1 induced the CD133−
cancer cells dedifferentiation in pancreatic cancer following radiotherapy. This multi-activity of extracellular HMGB1 in the regulation of the cancer cell biology may depend on its
secretory manner, redox status, cleavage, and special binding receptors. Our results also confirmed that both HMGB1 redox state and oxidized state promotes CD133−cancer cells
dedifferentiation (Fig. S6). Knockdown HMGB1 in the feeder cancer cells can not completely abrogate this effect. This can be ascribed to: (i) some other inflammatory factors (such as IL-1β
and TNF-a) released from the irradiation cell to the culture media have the similar function; (ii) FACS-purified cell population is not 100% pure. Indeed, we found sorted CD133− cancer cells
always contained a very small population of contaminating CD133+ cancer cells. YAP is overexpressed in many cancers and plays important roles in cancer cell proliferation, metabolism, and
treatment resistance26. Ectopic expression of YAP has been recently associated with cancer cell plasticity. Panciera et al. found that transient expression of YAP could converts
differentiated cells (neurons and pancreatic exocrine cells) into cells displaying multiple features of their corresponding tissue-specific stem cells27. In breast cancer, YAP/TAZ activity
is tightly linked with reprogram non-cancer stem cells into CSCs28. In hepatocellular carcinoma, elevated YAP activity was a key step conferring cancer cell with stem-like properties,
including chemoresistance and tumorigenicity, under continuous 5-FU treatment29. In this study, we found that dying cells-released HMGB1 promoted pancreatic cancer cells dedifferentiation by
decreasing YAP phosphorylation and triggering YAP nuclear translocation. Furthermore, YAP knockdown significantly decreased HIF-1α expression and inhibited HMGB1/TLR2 induced CD133− cancer
cell dedifferentiation. YAP mainly dependent on multiple domains to interact with transcription factors and form a complex to promote the expression and activation of downstream target genes
in the nucleus. HIF-1α could be activated in a hypoxia dependent and independent manner following radiotherapy30. Recent studies have demonstrated that HIF-1α was required for the
maintenance of CSCs in response to hypoxia or chemotherapy31,32. Dai et al. also reported that HIF-1α did not mediate hypoxia-triggered YAP nuclear translocation in hepatocellular carcinoma
cells33. Zhang et al. shown that YAP interacts directly with HIF-1α in the nucleus and sustains HIF-1α stability, which further correlated positively with hepatocellular carcinoma
progression. Our data are in line with the later observations to show that HMGB1 triggered both YAP and HIF-1α nuclear translocation and enhanced YAP and HIF-1α interaction. As silencing YAP
decreased HIF-1α protein expression, it is possible that YAP is important for HIF-1α stability in hypoxia independent manner under HMGB1/TLR2 treatment. Moreover, silencing HIF-1α decreased
the YAP nuclear translocation. Thus, HIF-1α is required for YAP performing the transcription factor functional under HMGB1/TLR2 treatment. Recently several studies reported that Nanog was
sufficient to confer cancer cells certain CSC properties and phenotypes in prostate and brain cancer34,35. From our in vitro study, it was obviously found that Nanog is the most obviously
changed gene in the process of CD133− pancreatic cancer cells dedifferentiation compare to the other target gene. In this study, we found that HMGB1-TLR2 mediated cytoplasmic YAP
translocation into the nucleus. Consequently, YAP and HIF-1α form complex in the nucleus and further induce CD133− cancer cells dedifferentiation. Based on our results, direct inducing
cancer cell death or targeting of CSCs may not be sufficient to cure cancer. Tumor microenvironmental, such as the paracrine signaling loops between dying cancer cells and resident
differentiated cancer cells, could represent the basis of new, innovative treatments target. Overall, we propose a novel strategy combining HMGB1 inhibitors with irradiation for synergistic
pancreatic carcinoma treatment. REFERENCES * Siegel, R., Ma, J., Zou, Z. & Jemal, A. Cancer statistics, 2014. _CA Cancer J. Clin._ 67, 7 (2014). Article Google Scholar * Barker, H. E.,
Paget, J. T., Khan, A. A. & Harrington, K. J. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. _Nat. Rev. Cancer_ 15, 409–425 (2015). Article
CAS Google Scholar * Landau, E. & Kalnicki, S. The evolving role of radiation in pancreatic cancer. _Surg. Clin. North Am._ 98, 113 (2018). Article Google Scholar * Ng, W. L. et al.
Molecular mechanisms involved in tumor repopulation after radiotherapy. _Transl. Cancer Res._ 2, 442–448 (2013). CAS PubMed PubMed Central Google Scholar * Ishiwata, T. et al. Pancreatic
cancer stem cells: features and detection methods. _Pathol. Oncol. Res._ 24, 797–805 (2018). Article CAS Google Scholar * Lagadec, C. et al. Survival and self-renewing capacity of breast
cancer initiating cells during fractionated radiation treatment. _Breast Cancer Res._ 12, R13 (2010). Article Google Scholar * Kiera, R. & Tang, D. G. Cancer stem cells and
radioresistance. _Int. J. Radiat. Biol._ 90, 615–621 (2014). Article Google Scholar * Kyjacova, L. et al. Radiotherapy-induced plasticity of prostate cancer mobilizes stem-like
non-adherent, Erk signaling-dependent cells. _Cell Death Differ._ 22, 898–911 (2015). Article CAS Google Scholar * Vlashi, E. et al. Radiation-induced dedifferentiation of head and neck
cancer cells into cancer stem cells depends on human papillomavirus status. _Int. J. Radiat. Oncol. Biol. Phys._ 94, 1198–1206 (2016). Article Google Scholar * Chann, L., Erina, V.,
Lorenza, D. D., Carmen, D. & Frank, P. Radiation-induced reprogramming of breast cancer cells. _Stem Cells_ 30, 833–844 (2012). Article Google Scholar * Dimitrios, I., Hirsch, H. A.,
Guannan, W. & Kevin, S. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. _Proc. Natl Acad. Sci. USA_ 108,
1397–1402 (2011). Article Google Scholar * Zhang, H. et al. Transforming growth factor β1 signal is crucial for dedifferentiation of cancer cells to cancer stem cells in osteosarcoma.
_Stem Cells_ 31, 433–446 (2013). Article Google Scholar * Rui, K., Qiuhong, Z., Zeh, H. J., Lotze, M. T. & Daolin, T. HMGB1 in cancer: good, bad, or both? _Clin. Cancer Res._ 19,
4046–4057 (2013). Article Google Scholar * Luo, Y. et al. High mobility group box 1 released from necrotic cells enhances regrowth and metastasis of cancer cells that have survived
chemotherapy. _Eur. J. Cancer_ 49, 741–751 (2013). Article CAS Google Scholar * He, S. et al. HMGB1 released by irradiated tumor cells promotes living tumor cell proliferation via
paracrine effect. _Cell Death Dis._ 9, 648 (2018). Article Google Scholar * Chen, X. et al. Radiotherapy-induced cell death activates paracrine HMGB1-TLR2 signaling and accelerates
pancreatic carcinoma metastasis. _J. Exp. Clin. Cancer Res._ 37, 77 (2018). Article Google Scholar * Haiyan, Z., Heppner, F. L. & Tsirka, S. E. Microglia/macrophages promote glioma
progression. _Glia_ 59, 472–485 (2011). Article Google Scholar * Zhu et al. Role of the hypoxia-inducible factor-1 alpha induced autophagy in the conversion of non-stem pancreatic cancer
cells into CD133(+) pancreatic;cancer stem-like cells. _Cancer Cell Int._ 13, 119 (2013). Article Google Scholar * Zhang, L. et al. CCL21/CCR7 axis contributed to CD133+ pancreatic cancer
stem-like cell metastasis via EMT and Erk/NF-κB pathway. _Plos ONE_ 11, e0158529 (2016). Article Google Scholar * Huang, C. Y. et al. Cytosolic high-mobility group box protein 1 (HMGB1)
and/or PD-1+ TILs in the tumor microenvironment may be contributing prognostic biomarkers for patients with locally advanced rectal cancer who have undergone neoadjuvant chemoradiotherapy.
_Cancer Immunol. Immunother._ 67, 551–562 (2018). Article CAS Google Scholar * Yin, H. et al. HMGB1-mediated autophagy attenuates gemcitabine-induced apoptosis in bladder cancer cells
involving JNK and ERK activation. _Oncotarget_ 8, 71642–71656 (2017). PubMed PubMed Central Google Scholar * Zhao, X. L. et al. High-mobility group box 1 released by autophagic
cancer-associated fibroblasts maintains the stemness of luminal breast cancer cells. _J. Pathol._ 243, 376–389 (2017). Article CAS Google Scholar * Lingyin, Z., Xiaobo, L., Yingxuan, C.,
Jingyuan, F. & Zhizheng, G. High-mobility group box 1: a novel inducer of the epithelial-mesenchymal transition in colorectal carcinoma. _Cancer Lett._ 357, 527–534 (2015). Article
Google Scholar * Scheel, C. & Weinberg, R. A. Cancer stem cells and epithelial–mesenchymal transition: concepts and molecular links. _Semin. Cancer Biol._ 22, 396–403 (2012). Article
CAS Google Scholar * Toshiro, M., Carsten Gram, H. & Kun-Liang, G. The emerging roles of YAP and TAZ in cancer. _Nat. Rev. Cancer_ 15, 73–79 (2015). Article Google Scholar *
Panciera, T. et al. Induction of expandable tissue-specific stem/progenitor cells through transient expression of YAP/TAZ. _Cell Stem Cell_ 19, 725–737 (2016). Article CAS Google Scholar
* Cordenonsi, M. et al. The hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. _Cell_ 147, 759–772 (2011). Article CAS Google Scholar * Hiromitsu, H. et
al. An imbalance in TAZ and YAP expression in hepatocellular carcinoma confers cancer stem cell-like behaviors contributing to disease progression. _Cancer Res._ 75, 4985–4997 (2016). Google
Scholar * Moeller, B. J., Cao, Y. C. & Dewhirst, M. W. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors; Role of reoxygenation, free radicals, and stress
granules. _Cancer Cell_ 5, 429–441 (2004). Article CAS Google Scholar * Samanta, D., Gilkes, D. M., Chaturvedi, P., Xiang, L. & Semenza, G. L. Hypoxia-inducible factors are required
for chemotherapy resistance of breast cancer stem cells. _Proc. Natl. Acad. Sci. USA_ 111, E5429–E5438 (2014). Article CAS Google Scholar * Dai, X. Y. et al. Nuclear translocation and
activation of YAP by hypoxia contributes to the chemoresistance of SN38 in hepatocellular carcinoma cells. _Oncotarget_ 7, 6933–6947 (2016). PubMed PubMed Central Google Scholar *
Haiquan, L. et al. Chemotherapy triggers HIF-1-dependent glutathione synthesis and copper chelation that induces the breast cancer stem cell phenotype. _Proc. Natl Acad. Sci. USA_ 112,
4600–4609 (2015). Article Google Scholar * Zhang, X. et al. Yes-associated protein (YAP) binds to HIF-1alpha and sustains HIF-1alpha protein stability to promote hepatocellular carcinoma
cell glycolysis under hypoxic stress. _J. Exp. Clin. Cancer Res._ 37, 216 (2018). Article Google Scholar * Jeter, C. R. et al. NANOG promotes cancer stem cell characteristics and prostate
cancer resistance to androgen deprivation. _Oncogene_ 30, 3833–3845 (2011). Article CAS Google Scholar * Machida, K. et al. Toll-like receptor 4 mediates synergism between alcohol and HCV
in hepatic oncogenesis involving stem cell marker Nanog. _Proc. Natl Acad. Sci. USA_ 106, 1548–1553 (2009). Article CAS Google Scholar Download references ACKNOWLEDGEMENTS This study was
supported by grants from the National Natural Science Foundation of China (Grant no. 81502663), the Social Development Foundation of Jiangsu Province (Grant no. BE2018691), Young medical
talents of Jiangsu (QNRC2016831, QNRC2016839), Six talent peals project of Jiangsu Province(WSW-039), the Social Development Foundation of Zhenjiang City (SH2017027, SH2018063), and Jiangsu
University Affiliated Hospital Startup fund for Ph.D. (jdfyrc2017010). AUTHORS’ CONTRIBUTIONS H.Z. and D.W. were corresponding authors and organized the study. L.Z., H.S., and X.X. performed
experiments. A.G., H.C., and Y.L. analyzed data. X.F. collected the clinical tumor tissue samples. T.Y. performed the radiotherapy. F.C. drafted the manuscript. All authors read and
approved the final manuscript. AUTHOR INFORMATION Author notes * These authors contributed equally: Lirong Zhang, Hui Shi, Hongbo Chen AUTHORS AND AFFILIATIONS * The Affiliated Hospital of
Jiangsu University, 212001, Zhenjiang, China Lirong Zhang, Hui Shi, Lian Song, Xuewen Xu, Tao You, Xin Fan, Dongqing Wang, Fang Cheng & Haitao Zhu * School of Pharmaceutical Sciences
(Shenzhen), SYSU, 518107, Shenzhen, China Hongbo Chen & Fang Cheng * School of Medicine, Jiangsu University, 212013, Zhenjiang, China Aihua Gong * The First People’s Hospital of
Zhenjiang, 212001, Zhenjiang, China Yanfang Liu * Faculty of Science and Engineering, ÅboAkademi University and Turku Centre for Biotechnology, FI-20520, Turku, Finland Fang Cheng Authors *
Lirong Zhang View author publications You can also search for this author inPubMed Google Scholar * Hui Shi View author publications You can also search for this author inPubMed Google
Scholar * Hongbo Chen View author publications You can also search for this author inPubMed Google Scholar * Aihua Gong View author publications You can also search for this author inPubMed
Google Scholar * Yanfang Liu View author publications You can also search for this author inPubMed Google Scholar * Lian Song View author publications You can also search for this author
inPubMed Google Scholar * Xuewen Xu View author publications You can also search for this author inPubMed Google Scholar * Tao You View author publications You can also search for this
author inPubMed Google Scholar * Xin Fan View author publications You can also search for this author inPubMed Google Scholar * Dongqing Wang View author publications You can also search for
this author inPubMed Google Scholar * Fang Cheng View author publications You can also search for this author inPubMed Google Scholar * Haitao Zhu View author publications You can also
search for this author inPubMed Google Scholar CORRESPONDING AUTHORS Correspondence to Dongqing Wang, Fang Cheng or Haitao Zhu. ETHICS DECLARATIONS CONFLICT OF INTEREST The authors declare
that they have no conflict of interest. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional
affiliations. Edited by M. Agostini SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0
International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the
source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Zhang, L., Shi, H., Chen, H. _et al._ Dedifferentiation process driven by
radiotherapy-induced HMGB1/TLR2/YAP/HIF-1α signaling enhances pancreatic cancer stemness. _Cell Death Dis_ 10, 724 (2019). https://doi.org/10.1038/s41419-019-1956-8 Download citation *
Received: 04 December 2018 * Revised: 16 August 2019 * Accepted: 04 September 2019 * Published: 26 September 2019 * DOI: https://doi.org/10.1038/s41419-019-1956-8 SHARE THIS ARTICLE Anyone
you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the
Springer Nature SharedIt content-sharing initiative